Showing 102 of 102 publications
2025 (14)
-

Lymphotoxin-driven cancer cell eradication by tumoricidal CD8(+) TIL
bioRxiv. 2025 Nov 20.
Abstract
Tumor-infiltrating lymphocyte (TIL) therapy is FDA-approved for patients with treatment-resistant advanced melanoma, but the TIL subpopulations critical for tumor eradication remains incompletely understood. Using patient-derived TIL-melanoma co-cultures, we identified and characterized a novel subset of CD8(+) TIL, capable of class I HLA-independent cancer cell lysis. The lymphotoxin β receptor (LTβR) and interferon (IFN) sensing pathways were nominated as key determinants of TIL-mediated cancer cell killing from a whole-genome, loss-of-function CRISPR screen. Validation studies confirmed that dual LTβR and IFN sensing is necessary and sufficient for cancer cell lysis, and that expanded CD8(+) TIL express high lymphotoxin β (LTB) and upregulate lymphotoxin α (LTA) upon coculture with cancer cells. Leveraging paired scRNA-seq and scTCR-seq data, we confirmed that enrichment of LTB (+) CD8 (+) T cells is associated with clinical response to TIL, and that LTB (+) CD8 (+) TIL are expanded from putative neoantigen-reactive, LTB (lo) CD8(+) T cells in resected tumors.
-
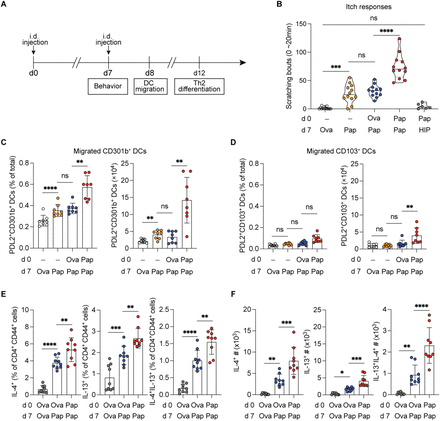
Sensory neuronal mTORC1 signaling establishes neuroimmune memory that initiates allergic immunity
bioRxiv. 2025 Nov 12.
Abstract
Environmental allergens are enriched in protease activity, a feature that directly activates cutaneous sensory neurons, triggering itch and Substance P release, which promotes Th2-skewing CD301b (+) dendritic cell migration and initiates allergic immunity. However, allergens are typically encountered as repeated, subthreshold exposures, and how these cumulatively induce sensitization is unknown. We identify a sensory neuron-intrinsic mechanism of neuroimmune memory that enables this process. Initial activation by protease allergens induces sustained mTORC1 signaling and PGC-1α-associated mitochondrial remodeling, establishing a metabolically primed state that amplifies neuroimmune responses upon allergen re-exposure. This heightened state, independent of adaptive immunity, drives enhanced itch, increased CD301b (+) dendritic cell migration, and augmented Th2 differentiation. Disrupting neuronal mTORC1 signaling or mitochondrial stability abrogates this amplification while leaving primary responses intact. Importantly, this mechanism generalizes across distinct protease allergens, revealing mTORC1-driven metabolic reprogramming in sensory neurons as a form of innate neuroimmune memory underlying allergen cross-sensitization and polysensitization.
-
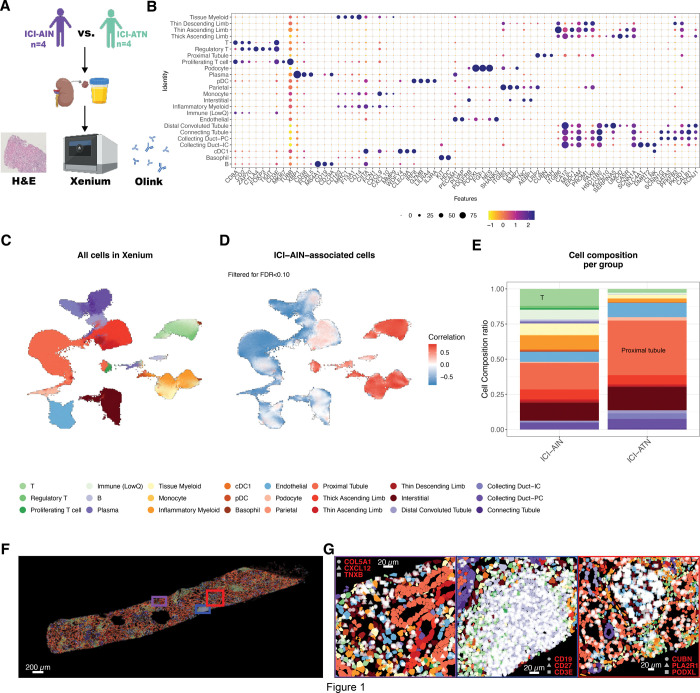
Spatial Transcriptomics Identify T Cell-Driven Mechanisms of Kidney Damage in Immune Checkpoint Inhibitor-Associated Acute Interstitial Nephritis
bioRxiv. 2025 Nov 3.
Abstract
INTRODUCTION: Immune checkpoint inhibitor-associated acute interstitial nephritis (ICI-AIN) is the most common finding on histopathology among patients with ICI-associated acute kidney injury (ICI-AKI). Patients with ICI-AIN often have T cell-dominant infiltration of the kidney and high tissue levels of CXCR3 ligands like CXCL9, 10, and 11; however, the mechanisms of inflammation in ICI-AIN are not well-understood. METHODS: We applied a sub-cellular spatial transcriptomics platform (Xenium Prime 5K) to compare the cellular composition of kidney biopsy tissue from patients with ICI-AIN with ICI-treated patients with acute tubular necrosis (ICI-ATN). RESULTS: Across 8 kidney biopsy specimens (4 with ICI-AIN, 4 with ICI-ATN), we analyzed 332,000 cells, comprising kidney parenchymal cells and infiltrating immune cells. Using a spatially-aware cellular neighborhood-based classification, we identified cellular niches corresponding to each part of the nephron, in addition to unique fibrotic and inflammatory niches. Gene pathway analysis identified interferon-gamma (IFN-γ)/STAT1 signaling as strongly increased in ICI-AIN compared to ICI-ATN. While all inflammatory niches were overrepresented in ICI-AIN, CD8(+) T cell infiltration and proinflammatory myeloid cells were the dominant immune niches. Spatial niche crosstalk analysis revealed that CD8(+) T cell-derived IFN-γ likely induced a proinflammatory program in myeloid cells, with increased production of CXCL9, 10, and 11. Furthermore, IFN-γ signaling in ICI-AIN was associated with reduced oxidative phosphorylation in kidney tubular niches. CONCLUSIONS: Spatial transcriptomics reveal novel insights into key differences in the pathophysiology of ICI-AIN versus ICI-ATN. IFN-γ-producing CD8(+) T cells are likely key drivers of ICI-AIN and should be investigated as future therapeutic targets.
-
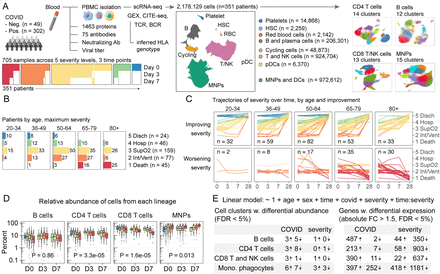
A multimodal atlas of COVID-19 severity identifies hallmarks of dysregulated immunity
medRxiv. 2025 Sep 21.
Abstract
The alpha-variant wave of the COVID-19 pandemic provided a unique opportunity to study, at single-cell resolution, how near-universal exposure to the same pathogen can lead to either effective or dysfunctional immune responses in humans. Although single-cell RNA-sequencing studies have characterized immune cellular features of COVID-19, they have not shown how tocilizumab treatment changes these features at single-cell resolution, or which features might persist into convalescence. In this study, we analyzed 2.5 million circulating immune cells from 428 patients across time points (840 PBMC samples), encompassing three contemporaneous SARS-CoV-2 cohorts: acutely infected patients across five WHO disease severity levels and three time points, patients from the first randomized control trial to study the efficacy of tocilizumab in the management of COVID-19, and convalescent patients three months after infection. We used linear modeling to integrate multiple data types - including single-cell RNA-seq, CITE-seq, TCR and BCR sequencing, viral load measurements, viral neutralization assays, detection of 75 autoantibodies, HLA genotype data, and serum proteomics covering 1,463 targets - to derive the most comprehensive view to-date of the biological features of COVID-19 disease severity. Our findings show that myeloid-derived suppressor cells (MDSCs) act as a key immunologic pivot point in severe COVID-19. Myeloid dysfunction, which is marked by impaired antigen presentation, drives a non-productive adaptive immune response, as reflected by reduced expression of B and T cell gene programs involved in antigen recognition, immune synapse formation, and cytotoxicity. Severe disease is also linked to autoantibodies targeting type I interferons, influenced by specific HLA-DQB1 allelic variants, and strongly correlated with serum IL-6 levels. Tocilizumab treatment eliminates CLU -expressing MDSCs and ISG-positive myeloid subsets, restores antigen presentation, and reactivates productive adaptive immunity. These changes align with improved clinical outcomes and better clinical laboratory measures, including reduced CRP. While many immunologic abnormalities in acute severe COVID-19 resolve during convalescence 3-months post-infection, we observed persistently high ICOS expression in regulatory T cells, potentially linking acute infection to chronic post-COVID syndromes. Overall, we define distinct innate and adaptive host immune responses associated with acute, IL-6-responsive, and convalescent SARS-CoV-2 infection. Our multimodal and high-dimensional dataset with curated clinical metadata provides a foundational and clinically relevant resource for modeling host immune response biology in health and disease.
-
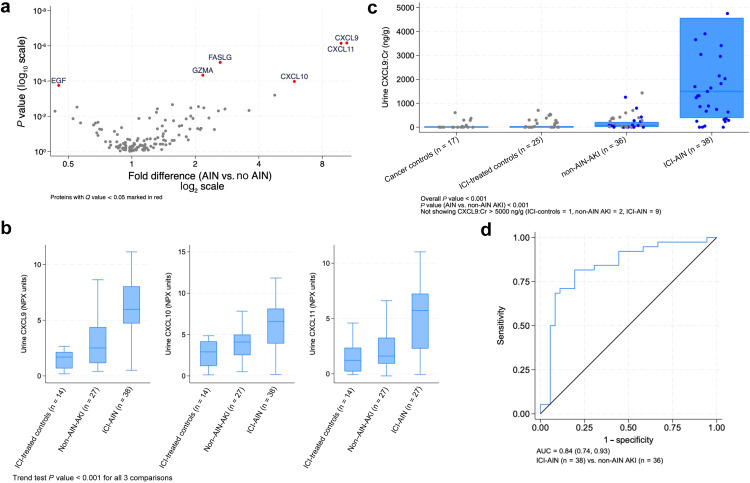
Urinary C-X-C-motif ligand 9 (CXCL9) in immune checkpoint inhibitor-associated acute interstitial nephritis
Kidney Int, 108(3):491-496. 2025 Sep.
Abstract
INTRODUCTION: Immune checkpoint inhibitor-associated acute interstitial nephritis presents significant clinical challenges. There are no reliable non-invasive biomarkers and kidney biopsy remains the gold standard for diagnosis. Prior studies have shown that urinary C-X-C-motif ligand 9 (CXCL9) is upregulated in patients with acute interstitial nephritis. However, its utility, specifically in patients with cancer treated with immune checkpoint inhibitors, is not well-understood. METHODS: We used proteomics followed by sandwich immunoassay to analyze urinary proteins among a multicenter cohort of prospectively enrolled participants with and without immune checkpoint inhibitor-associated acute interstitial nephritis. RESULTS: Among 79 participants receiving immune checkpoint inhibitors, proteomics identified urine CXCL9 as the top-performing urinary biomarker differentiating 38 patients with biopsy-proven acute interstitial nephritis from other forms of acute kidney injury. We validated these results using immunoassay in an expanded cohort of 116 patients, observing higher CXCL9 levels in immune checkpoint inhibitor-associated acute interstitial nephritis compared to several control groups. Urinary CXCL9 was strongly associated with immune checkpoint inhibitor-associated acute interstitial nephritis, with a receiver operating characteristic curve of 0.84, interquartile range [0.74, 0.93] when compared to other forms of acute kidney injury, and an even higher discrimination when compared with all control groups (0.90, [0.83-0.96]). CONCLUSIONS: Urinary CXCL9 demonstrated high discrimination for differentiating acute interstitial nephritis from other forms of acute kidney injury in participants on immune checkpoint inhibitor therapy. Our findings demonstrate the significant potential of this biomarker for non-invasive diagnosis of immune checkpoint inhibitor-associated acute interstitial nephritis.
-

Airway basal stem cells are necessary for the maintenance of functional intraepithelial airway macrophages
Cell Rep, 44(6):115860. 2025 Jun 24.
Abstract
Stem cells are known to provide signals that contribute to the maintenance and function of neighboring cells. We demonstrate that Notch signaling arising from airway basal stem cells is necessary for the function of a unique population of intraepithelial airway macrophages (IAMs) in the murine trachea. Without this stem cell signaling, IAMs lose MHC II expression, which in turn prevents antigen-induced allergic inflammation. Distal murine airways do not harbor basal stem cells, and, in this region of the lung, allergic inflammation proceeds unperturbed. We speculate that the functional coupling of specific anatomically restricted stem cell populations and adjacent immune cells is one mechanism for ensuring that inflammatory responses are compartmentalized to regions of injury. Basal stem cells are found throughout the human airway tree and we demonstrate the existence of human IAM-like cells, suggesting that their interaction may influence airways disease.
-
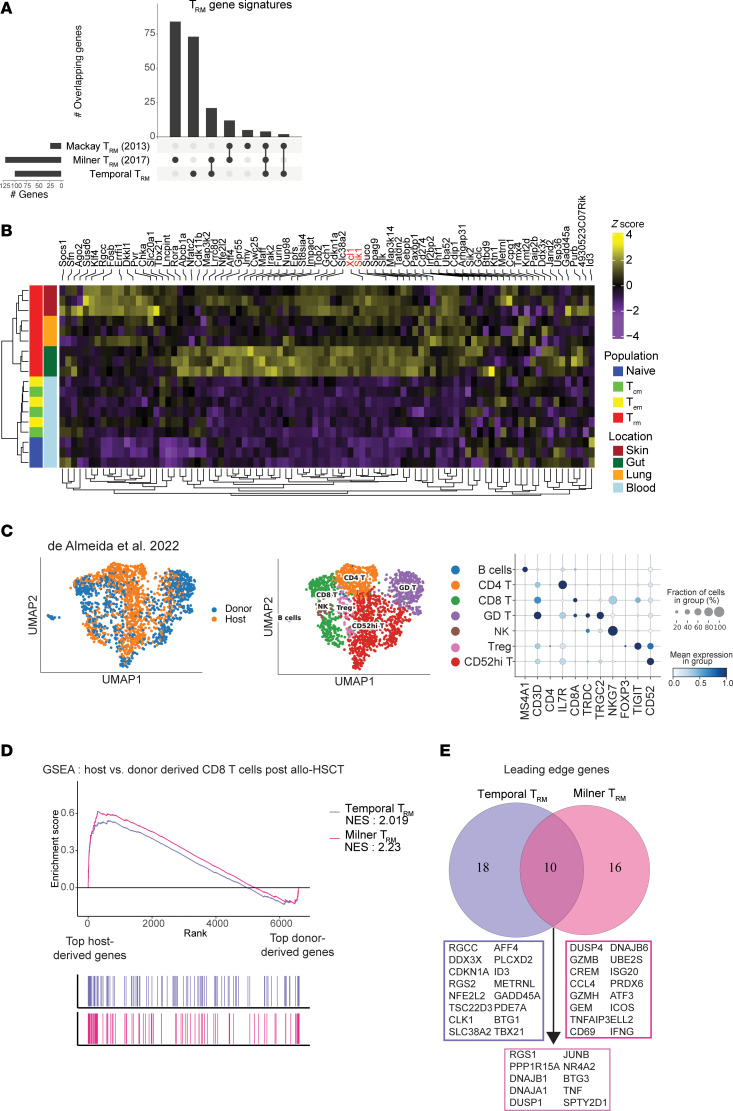
Resident memory T cell development is gradual and shows AP-1 gene expression in mature cells
JCI Insight, 10(12). 2025 Jun 23.
Abstract
Tissue-resident memory T (TRM) cells play a central role in immune responses across all barrier tissues after infection. However, the mechanisms that drive TRM differentiation and priming for their recall effector function remains unclear. In this study, we leveraged newly generated and publicly available single-cell RNA-seq data generated across 10 developmental time points to define features of CD8+ TRM across both skin and small-intestine intraepithelial lymphocytes (siIEL). We employed linear modeling to capture gene programs that increase their expression levels in T cells transitioning from an effector to a memory state. In addition to capturing tissue-specific gene programs, we defined a temporal TRM signature across skin and siIEL that can distinguish TRM from circulating T cell populations. This TRM signature highlights biology that is missed in published signatures that compared bulk TRM to naive or nontissue resident memory populations. This temporal TRM signature included the AP-1 transcription factor family members Fos, Fosb, Fosl2, and Junb. ATAC-seq analysis detected AP-1-specific motifs at open chromatin sites in mature TRM. Cyclic immunofluorescence (CyCIF) tissue imaging detected nuclear colocalization of AP-1 members in resting CD8+ TRM greater than 100 days after infection. Taken together, these results reveal a critical role of AP-1 transcription factor members in TRM biology.
-
Single cell RNA sequencing shows that cells expressing Sox9 postnatally populate most skeletal lineages in mouse bone
J Bone Miner Res, 40(6):799-812. 2025 Jun 3.
Abstract
In growing bones of mice, multiple cell types contribute to the osteoblast lineage, including growth plate chondrocytes, perichondrial cells and CXCL12-abundant reticular marrow stromal cells. Here we use single-cell RNA sequencing and lineage tracing to show that all these osteoblast precursors, even postnatally, derives from Sox9-expressing progenitors. We also characterize a distinct group of chondrocytes located between the perichondrium and the columns of growth plate chondrocytes that contribute to the osteoblast lineage.
-

Diverse modes of T cell receptor sequence convergence define unique functional and cellular phenotypes
bioRxiv. 2025 Jun 3.
Abstract
Single-cell techniques allow concurrent study of gene activity and T cell receptor (TCR) sequences, identifying connections between TCR structure and cell traits. Expanding on our CoNGA software, we present a "metaCoNGA" analysis of 6 million T cells from 91 diverse studies, mapping TCR sequence similarity across tissues and diseases. This approach exposes shared TCR features within specific T cell subsets, including those associated with infection, cancer, and autoimmunity. We introduce a method to identify T cell groups with similar gene expression and biased TCR amino acid composition, providing a systematic framework for classifying diverse unconventional T cells, including KIR+ CD8+ T cells, CD4+ regulatory T cells, and subsets of NKT and MAIT cells. A new TCR clustering approach identifies thousands of convergent TCR sequence clusters hypothesized to target shared antigens. These clusters show coherent gene expression, highlighting the role of antigen exposure in shaping T cell behavior. Finally, we provide a tool for users to merge new data with this resource and rapidly identify T cell features in their data sets. This resource empowers investigations into the complex relationship between TCR sequence and T cell function in human health.
-
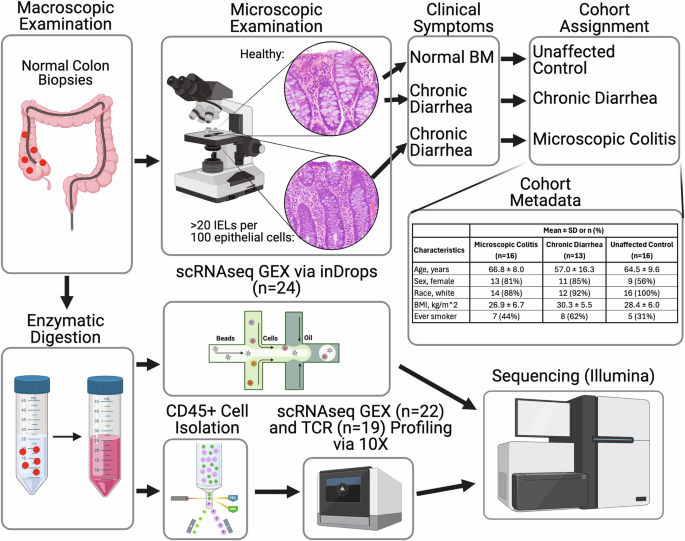
Single-cell transcriptomic characterization of microscopic colitis
Nat Commun, 16(1):4618. 2025 May 18.
Abstract
Microscopic colitis (MC) is a chronic inflammatory disease of the large intestine and a common cause of chronic diarrhea in older adults. Here, we use single-cell RNA sequencing analysis of colonic mucosal tissue to build a cellular and molecular model for MC. Our results show that in MC, there is a substantial expansion of tissue CD8(+) T cells, likely arising from local expansion following T cell receptor engagement. Within the T cell compartment, MC is characterized by a shift in CD8 tissue-resident memory T cells towards a highly cytotoxic and inflammatory phenotype and expansion of CD4(+) T regulatory cells. These results provide insight into inflammatory cytokines shaping MC pathogenesis and highlight notable similarities and differences with other immune-mediated intestinal diseases, including a common upregulation of IL26 and an MC-specific upregulation of IL10. These data help identify targets against enteric T cell subsets as an effective strategy for treatment of MC.
-
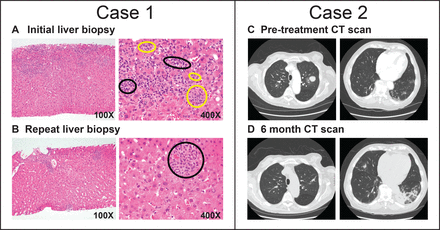
Tumor location as a risk factor for severe immune-related adverse events
J Immunother Cancer, 13(5). 2025 May 15.
Abstract
Immune-related adverse events (irAEs) can cause severe morbidity and mortality, and they impair treatment with immune checkpoint inhibitors (ICI). Risk factors for irAEs are not well understood.We observed cases of patients having tumor deposits in their liver and lung during a workup of irAEs, which led us to hypothesize that the presence of tumor in an organ would increase the odds of developing severe irAEs in that organ. We then performed a retrospective cohort study that included patients who received an ICI for the treatment of cancer and were hospitalized between February 2011 and November 2021 at the Massachusetts General Hospital.We reviewed 384 patients hospitalized with concern for any irAE. A clinical diagnosis of ICI-related hepatitis occurred in 18% of patients with liver tumor deposits versus 8% of those without (OR 2.23, 95% CI (1.10 to 4.43), p=0.02). ICI-related pneumonitis occurred in 10% of patients with lung tumor deposits versus 4.4% of those without (OR 2.45, 95% CI (1.06 to 6.36), p=0.047). A combined analysis for liver and lung lesions demonstrated that the presence of tumor deposits in an organ increased the odds of having an irAE in that organ by over twofold (OR 2.31, 95% CI (1.34 to 3.99), p=0.002).Our results suggest that the presence of tumor deposits may represent a novel risk factor for severe irAEs in that organ.
-
Asian diversity in human immune cells
Cell, 188(8):2288-2306.e24. 2025 Apr 17.
Abstract
The relationships of human diversity with biomedical phenotypes are pervasive yet remain understudied, particularly in a single-cell genomics context. Here, we present the Asian Immune Diversity Atlas (AIDA), a multi-national single-cell RNA sequencing (scRNA-seq) healthy reference atlas of human immune cells. AIDA comprises 1,265,624 circulating immune cells from 619 donors, spanning 7 population groups across 5 Asian countries, and 6 controls. Though population groups are frequently compared at the continental level, we found that sub-continental diversity, age, and sex pervasively impacted cellular and molecular properties of immune cells. These included differential abundance of cell neighborhoods as well as cell populations and genes relevant to disease risk, pathogenesis, and diagnostics. We discovered functional genetic variants influencing cell-type-specific gene expression, which were under-represented in non-Asian populations, and helped contextualize disease-associated variants. AIDA enables analyses of multi-ancestry disease datasets and facilitates the development of precision medicine efforts in Asia and beyond.
-
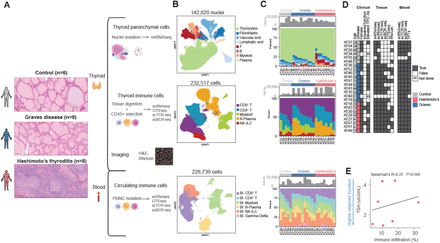
Immune-parenchymal multicellular niches are shared across distinct thyroid autoimmune diseases
bioRxiv. 2025 Apr 3.
Abstract
Thyroid hormone, produced in the thyroid gland, regulates metabolism, development, and cardiac function. The thyroid is susceptible to autoimmune attack by both cellular and humoral immunity exemplified by Hashimoto's thyroiditis (HT) and Graves' Disease (GD), respectively. In HT, immune-mediated destruction impairs thyroid hormone production, while in GD, stimulating autoantibodies promote over-production. Here, we generated a multi-modal atlas of 604,076 human thyroid and blood cells from HT, GD, and control patients. We found that, despite markedly different clinical presentations and distinct antigenic triggers, HT and GD exhibit convergent cellular dynamics resulting in a shared continuum of immune infiltration. Along this continuum, a key feature is a thyrocyte niche containing CD8 (+) T cells that may segregate pathogenic T cells from regions with preserved thyroid hormone production. These findings of a shared disease continuum characterized by spatially defined immune niches provide a new framework for understanding tissue homeostasis in human autoimmune disease.
-
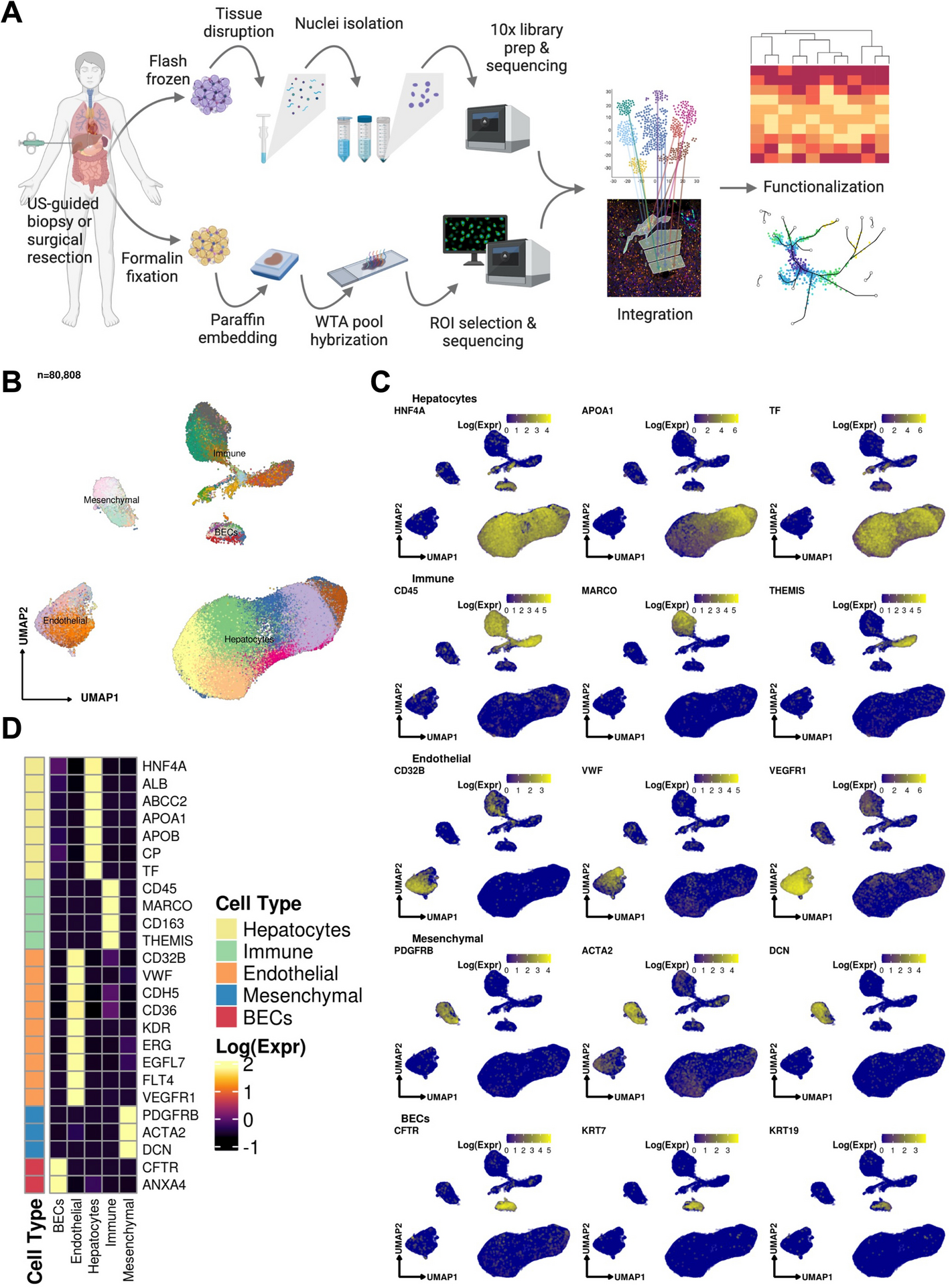
A single-nucleus and spatial transcriptomic atlas of the COVID-19 liver reveals topological, functional, and regenerative organ disruption in patients
Genome Biol, 26(1):56. 2025 Mar 14.
Abstract
BACKGROUND: The molecular underpinnings of organ dysfunction in severe COVID-19 and its potential long-term sequelae are under intense investigation. To shed light on these in the context of liver function, we perform single-nucleus RNA-seq and spatial transcriptomic profiling of livers from 17 COVID-19 decedents. RESULTS: We identify hepatocytes positive for SARS-CoV-2 RNA with an expression phenotype resembling infected lung epithelial cells, and a central role in a pro-fibrotic TGFβ signaling cell-cell communications network. Integrated analysis and comparisons with healthy controls reveal extensive changes in the cellular composition and expression states in COVID-19 liver, providing the underpinning of hepatocellular injury, ductular reaction, pathologic vascular expansion, and fibrogenesis characteristic of COVID-19 cholangiopathy. We also observe Kupffer cell proliferation and erythrocyte progenitors for the first time in a human liver single-cell atlas. Despite the absence of a clinical acute liver injury phenotype, endothelial cell composition is dramatically impacted in COVID-19, concomitantly with extensive alterations and profibrogenic activation of reactive cholangiocytes and mesenchymal cells. CONCLUSIONS: Our atlas provides novel insights into liver physiology and pathology in COVID-19 and forms a foundational resource for its investigation and understanding.
2024 (13)
-
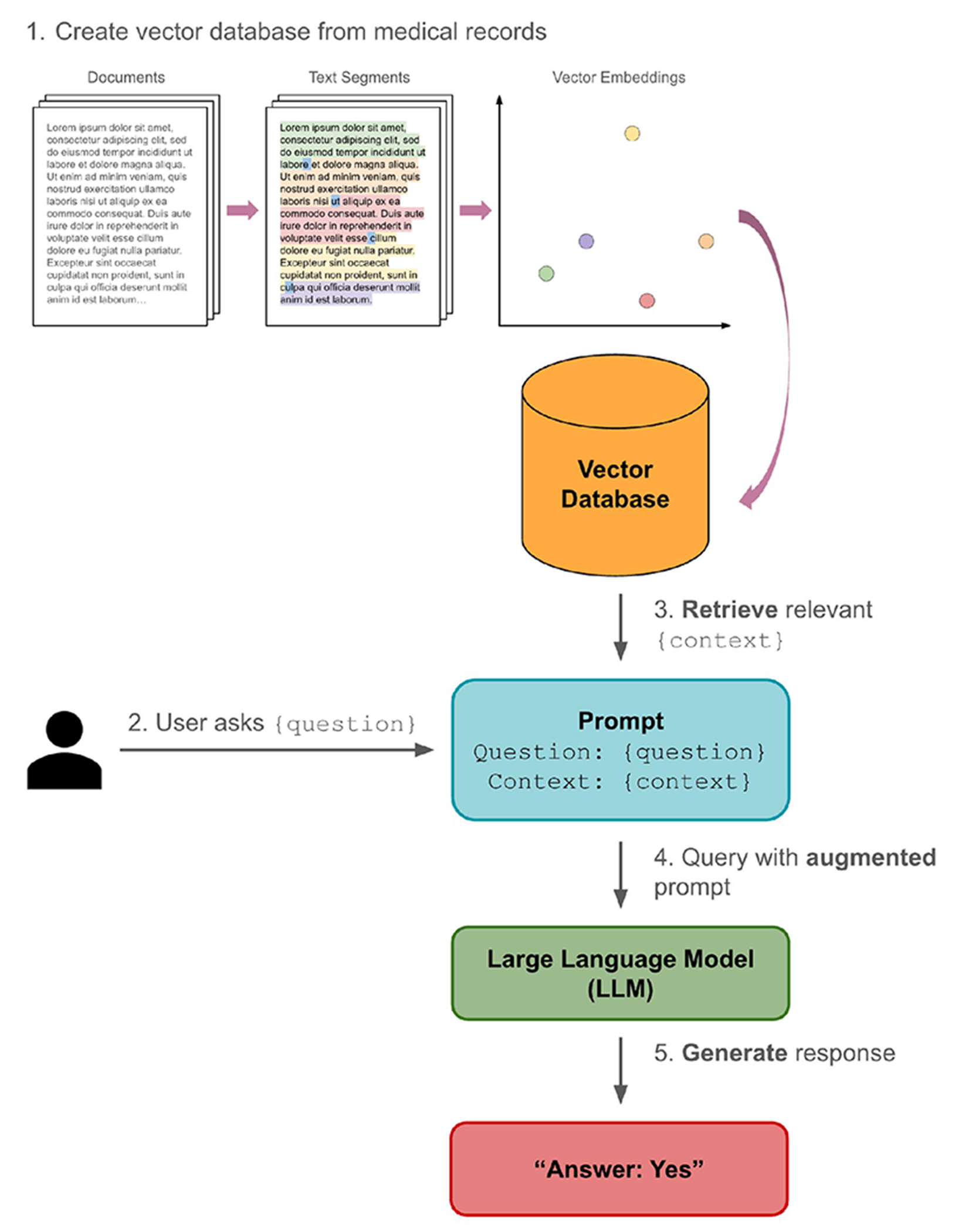
Enhancing Precision in Detecting Severe Immune-Related Adverse Events: Comparative Analysis of Large Language Models and International Classification of Disease Codes in Patient Records
J Clin Oncol, 42(35):4134-4144. 2024 Dec 10.
Abstract
PURPOSE: Current approaches to accurately identify immune-related adverse events (irAEs) in large retrospective studies are limited. Large language models (LLMs) offer a potential solution to this challenge, given their high performance in natural language comprehension tasks. Therefore, we investigated the use of an LLM to identify irAEs among hospitalized patients, comparing its performance with manual adjudication and International Classification of Disease (ICD) codes. METHODS: Hospital admissions of patients receiving immune checkpoint inhibitor (ICI) therapy at a single institution from February 5, 2011, to September 5, 2023, were individually reviewed and adjudicated for the presence of irAEs. ICD codes and an LLM with retrieval-augmented generation were applied to detect frequent irAEs (ICI-induced colitis, hepatitis, and pneumonitis) and the most fatal irAE (ICI-myocarditis) from electronic health records. The performance between ICD codes and LLM was compared via sensitivity and specificity with an α = .05, relative to the gold standard of manual adjudication. External validation was performed using a data set of hospital admissions from June 1, 2018, to May 31, 2019, from a second institution. RESULTS: Of the 7,555 admissions for patients on ICI therapy in the initial cohort, 2.0% were adjudicated to be due to ICI-colitis, 1.1% ICI-hepatitis, 0.7% ICI-pneumonitis, and 0.8% ICI-myocarditis. The LLM demonstrated higher sensitivity than ICD codes (94.7% v 68.7%), achieving significance for ICI-hepatitis (P < .001), myocarditis (P < .001), and pneumonitis (P = .003) while yielding similar specificities (93.7% v 92.4%). The LLM spent an average of 9.53 seconds/chart in comparison with an estimated 15 minutes for adjudication. In the validation cohort (N = 1,270), the mean LLM sensitivity and specificity were 98.1% and 95.7%, respectively. CONCLUSION: LLMs are a useful tool for the detection of irAEs, outperforming ICD codes in sensitivity and adjudication in efficiency.
-
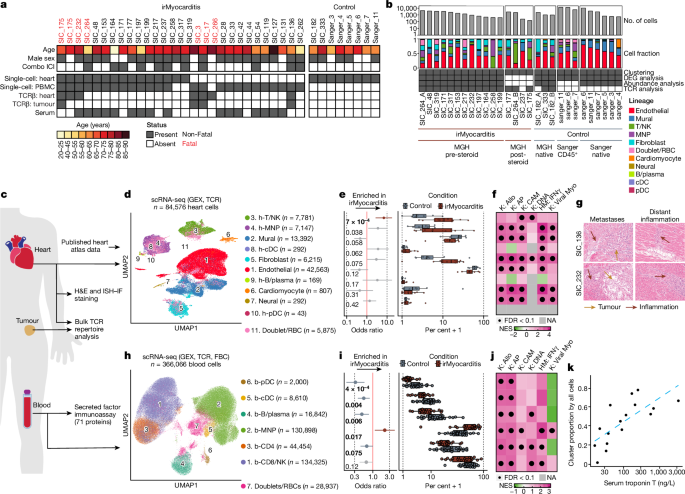
Immune responses in checkpoint myocarditis across heart, blood and tumour
Nature, 636(8041):215-223. 2024 Dec.
Abstract
Immune checkpoint inhibitors are widely used anticancer therapies(1) that can cause morbid and potentially fatal immune-related adverse events such as immune-related myocarditis (irMyocarditis)(2-5). The pathogenesis of irMyocarditis and its relationship to antitumour immunity remain poorly understood. Here we sought to define immune responses in heart, tumour and blood in patients with irMyocarditis by leveraging single-cell RNA sequencing coupled with T cell receptor (TCR) sequencing, microscopy and proteomics analyses of samples from 28 patients with irMyocarditis and 41 unaffected individuals. Analyses of 84,576 cardiac cells by single-cell RNA sequencing combined with multiplexed microscopy demonstrated increased frequencies and co-localization of cytotoxic T cells, conventional dendritic cells and inflammatory fibroblasts in irMyocarditis heart tissue. Analyses of 366,066 blood cells revealed decreased frequencies of plasmacytoid dendritic cells, conventional dendritic cells and B lineage cells but an increased frequency of other mononuclear phagocytes in irMyocarditis. Fifty-two heart-expanded TCR clones from eight patients did not recognize the putative cardiac autoantigens α-myosin, troponin I or troponin T. Additionally, TCRs enriched in heart tissue were largely nonoverlapping with those enriched in paired tumour tissue. The presence of heart-expanded TCRs in a cycling blood CD8 T cell population was associated with fatal irMyocarditis case status. Collectively, these findings highlight crucial biology driving irMyocarditis and identify putative biomarkers.
-
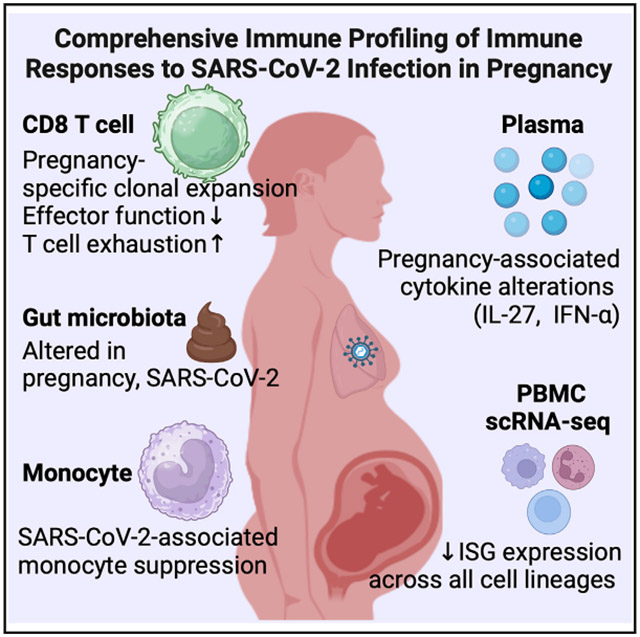
SARS-CoV-2 infection elucidates features of pregnancy-specific immunity
Cell Rep, 43(11):114933. 2024 Nov 26.
Abstract
Pregnancy is a risk factor for increased severity of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and other respiratory infections, but the mechanisms underlying this risk are poorly understood. To gain insight into the role of pregnancy in modulating immune responses at baseline and upon SARS-CoV-2 infection, we collected peripheral blood mononuclear cells and plasma from 226 women, including 152 pregnant individuals and 74 non-pregnant women. We find that SARS-CoV-2 infection is associated with altered T cell responses in pregnant women, including a clonal expansion of CD4-expressing CD8(+) T cells, diminished interferon responses, and profound suppression of monocyte function. We also identify shifts in cytokine and chemokine levels in the sera of pregnant individuals, including a robust increase of interleukin-27, known to drive T cell exhaustion. Our findings reveal nuanced pregnancy-associated immune responses, which may contribute to the increased susceptibility of pregnant individuals to viral respiratory infection.
-
The commitment of the human cell atlas to humanity
Nat Commun, 15(1):10019. 2024 Nov 20.
Abstract
The Human Cell Atlas (HCA) is a global partnership "to create comprehensive reference maps of all human cells-the fundamental units of life - as a basis for both understanding human health and diagnosing, monitoring, and treating disease." ( https://www.humancellatlas.org/ ) The atlas shall characterize cells from diverse individuals across the globe to better understand human biology. HCA proactively considers the priorities of, and benefits accrued to, contributing communities. Here, we lay out principles and action items that have been adopted to affirm HCA's commitment to equity so that the atlas is beneficial to all of humanity.
-
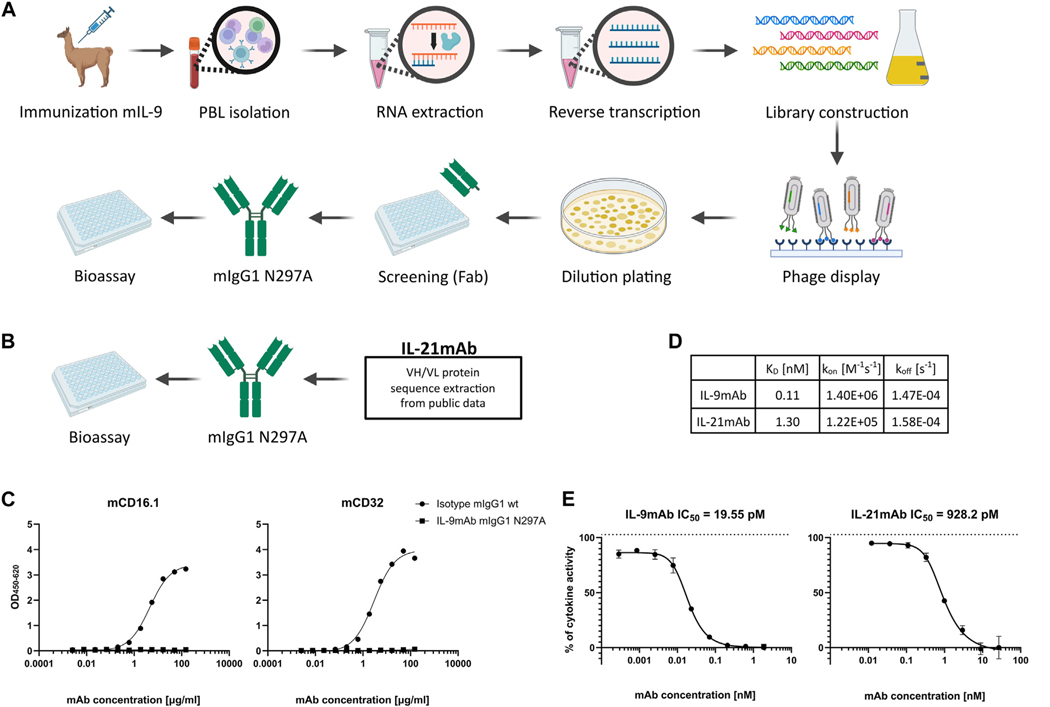
IL-2 family cytokines IL-9 and IL-21 differentially regulate innate and adaptive type 2 immunity in asthma
J Allergy Clin Immunol, 154(5):1129-1145. 2024 Nov.
Abstract
BACKGROUND: Asthma is often accompanied by type 2 immunity rich in IL-4, IL-5, and IL-13 cytokines produced by T(H)2 lymphocytes or type 2 innate lymphoid cells (ILC2s). IL-2 family cytokines play a key role in the differentiation, homeostasis, and effector function of innate and adaptive lymphocytes. OBJECTIVE: IL-9 and IL-21 boost activation and proliferation of T(H)2 and ILC2s, but the relative importance and potential synergism between these γ common chain cytokines are currently unknown. METHODS: Using newly generated antibodies, we inhibited IL-9 and IL-21 alone or in combination in various murine models of asthma. In a translational approach using segmental allergen challenge, we recently described elevated IL-9 levels in human subjects with allergic asthma compared with nonasthmatic controls. Here, we also measured IL-21 in both groups. RESULTS: IL-9 played a central role in controlling innate IL-33-induced lung inflammation by promoting proliferation and activation of ILC2s in an IL-21-independent manner. Conversely, chronic house dust mite-induced airway inflammation, mainly driven by adaptive immunity, was solely dependent on IL-21, which controlled T(H)2 activation, eosinophilia, total serum IgE, and formation of tertiary lymphoid structures. In a model of innate on adaptive immunity driven by papain allergen, a clear synergy was found between both pathways, as combined anti-IL-9 or anti-IL-21 blockade was superior in reducing key asthma features. In human bronchoalveolar lavage samples we measured elevated IL-21 protein within the allergic asthmatic group compared with the allergic control group. We also found increased IL21R transcripts and predicted IL-21 ligand activity in various disease-associated cell subsets. CONCLUSIONS: IL-9 and IL-21 play important and nonredundant roles in allergic asthma by boosting ILC2s and T(H)2 cells, revealing a dual IL-9 and IL-21 targeting strategy as a new and testable approach.
-
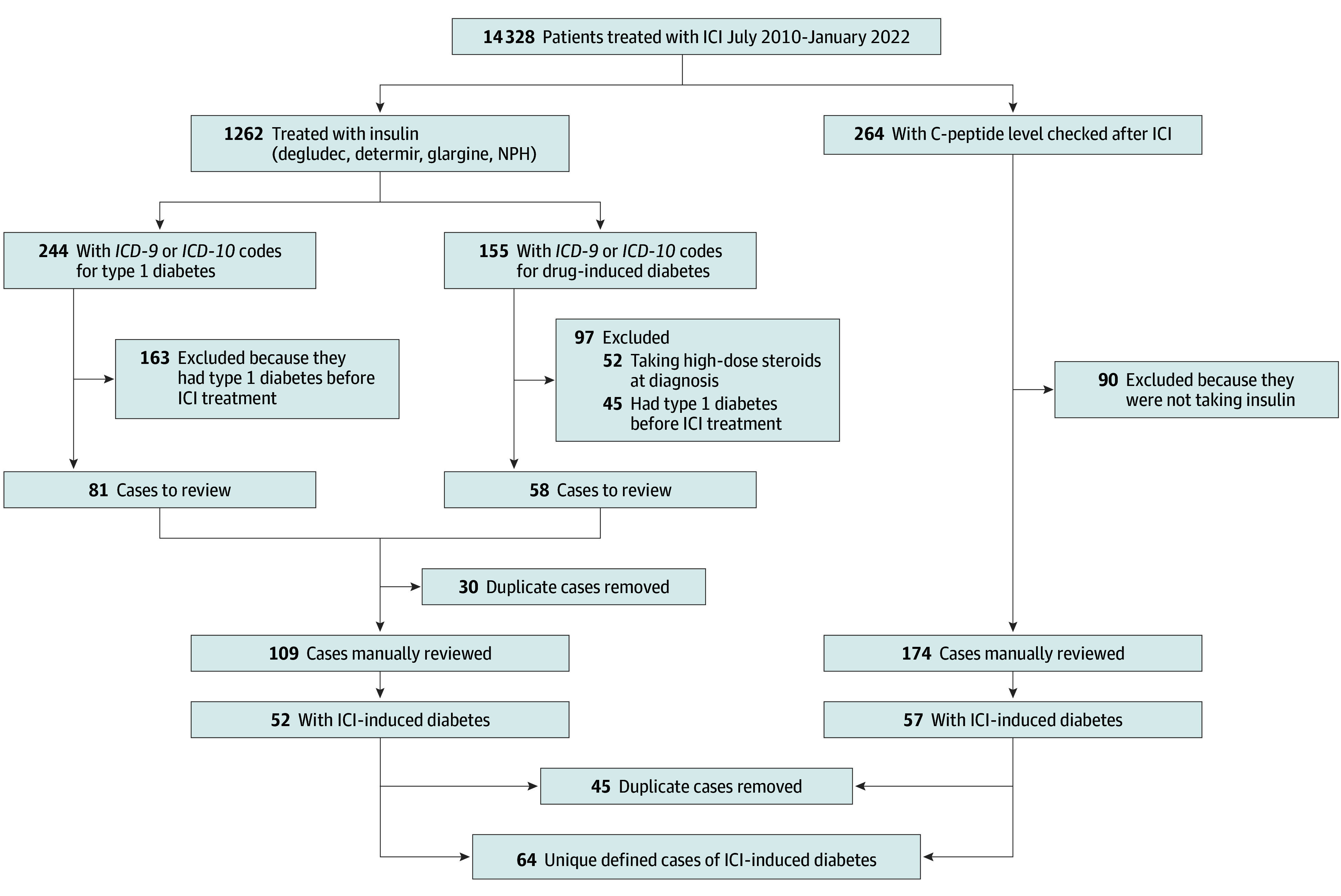
Identification of Immune Checkpoint Inhibitor-Induced Diabetes
JAMA Oncol, 10(10):1409-1416. 2024 Oct 1.
Abstract
IMPORTANCE: Immune checkpoint inhibitors (ICIs) have revolutionized cancer care; however, accompanying immune-related adverse events (irAEs) confer substantial morbidity and occasional mortality. Life-threatening irAEs may require permanent cessation of ICI, even in patients with positive tumor response. Therefore, it is imperative to comprehensively define the spectrum of irAEs to aid individualized decision-making around the initiation of ICI therapy. OBJECTIVE: To define incidence, risk factors, and clinical spectrum of an irreversible and life-threatening irAE: ICI-induced diabetes. DESIGN, SETTING, AND PARTICIPANTS: This cohort study, conducted at an academic integrated health care system examined 14 328 adult patients treated with ICIs, including 64 patients who developed ICI-induced diabetes, from July 2010 to January 2022. The data were analyzed from 2022 to 2023. Cases of ICI-induced diabetes were manually confirmed; detailed clinical phenotyping was performed at diagnosis and 1-year follow-up. For 862 patients, genotyping data were available, and polygenic risk for type 1 diabetes was determined. MAIN OUTCOMES AND MEASURES: For ICI-induced diabetes cases and controls, demographic characteristics, comorbidities, tumor category, and ICI category were compared. Among ICI-induced diabetes cases, markers of glycemic physiology were examined at diagnosis and 1-year follow-up. For patients with available genotyping, a published type 1 diabetes polygenic score (T1D GRS2) was calculated. RESULTS: Of 14 328 participants, 6571 (45.9%) were women, and the median (range) age was 66 (8-106) years. The prevalence of ICI-induced diabetes among ICI-treated patients was 0.45% (64 of 14 328), with an incidence of 124.8 per 100 000 person-years. Preexisting type 2 diabetes (odds ratio [OR], 5.91; 95% CI, 3.34-10.45) and treatment with combination ICI (OR, 2.57; 95% CI, 1.44-4.59) were significant clinical risk factors of ICI-induced diabetes. T1D GRS2 was associated with ICI-induced diabetes risk, with an OR of 4.4 (95% CI, 1.8-10.5) for patients in the top decile of T1D GRS2, demonstrating a genetic association between spontaneous autoimmunity and irAEs. Patients with ICI-induced diabetes were in 3 distinct phenotypic categories based on autoantibodies and residual pancreatic function, with varying severity of initial presentation. CONCLUSIONS AND RELEVANCE: The results of this analysis of 14 328 ICI-treated patients followed up from ICI initiation determined the incidence, risk factors and clinical spectrum of ICI-induced diabetes. Widespread implementation of this approach across organ-specific irAEs may enhance diagnosis and management of these conditions, and this becomes especially pertinent as ICI treatment rapidly expands to treat a wide spectrum of cancers and is used at earlier stages of treatment.
-
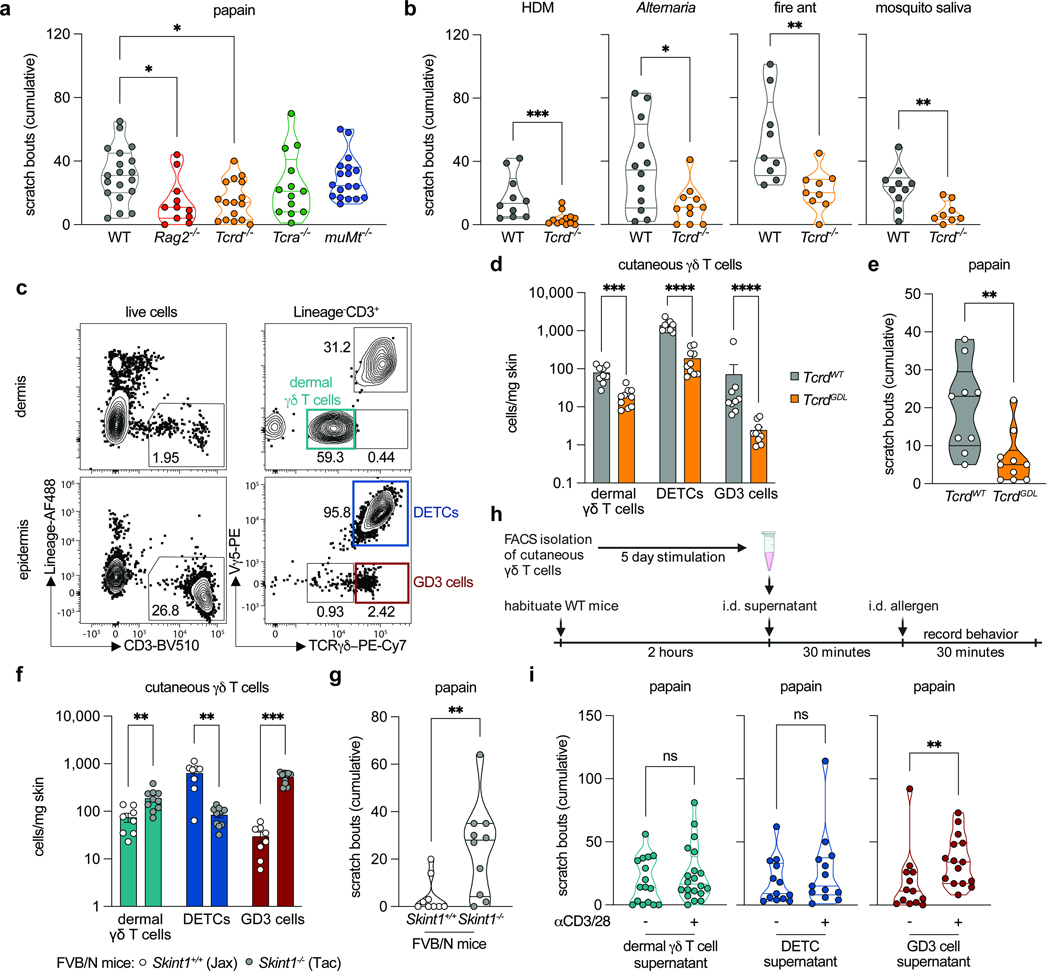
A γδ T cell-IL-3 axis controls allergic responses through sensory neurons
Nature, 634(8033):440-446. 2024 Oct.
Abstract
In naive individuals, sensory neurons directly detect and respond to allergens, leading to both the sensation of itch and the activation of local innate immune cells, which initiate the allergic immune response(1,2). In the setting of chronic allergic inflammation, immune factors prime sensory neurons, causing pathologic itch(3-7). Although these bidirectional neuroimmune circuits drive responses to allergens, whether immune cells regulate the set-point for neuronal activation by allergens in the naive state is unknown. Here we describe a γδ T cell-IL-3 signalling axis that controls the allergen responsiveness of cutaneous sensory neurons. We define a poorly characterized epidermal γδ T cell subset(8), termed GD3 cells, that produces its hallmark cytokine IL-3 to promote allergic itch and the initiation of the allergic immune response. Mechanistically, IL-3 acts on Il3ra-expressing sensory neurons in a JAK2-dependent manner to lower their threshold for allergen activation without independently eliciting itch. This γδ T cell-IL-3 signalling axis further acts by means of STAT5 to promote neuropeptide production and the initiation of allergic immunity. These results reveal an endogenous immune rheostat that sits upstream of and governs sensory neuronal responses to allergens on first exposure. This pathway may explain individual differences in allergic susceptibility and opens new therapeutic avenues for treating allergic diseases.
-
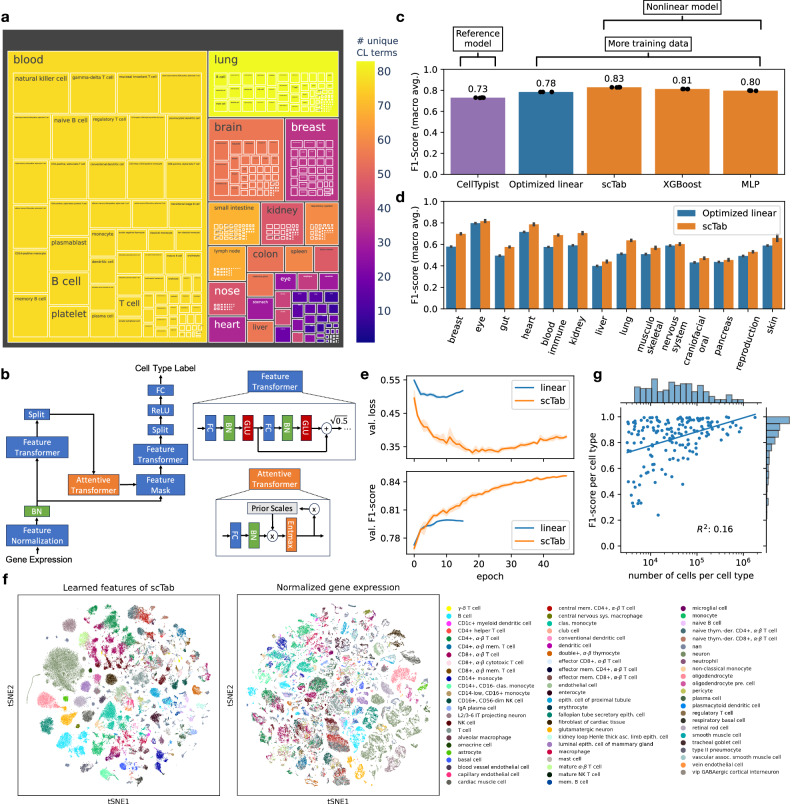
scTab: Scaling cross-tissue single-cell annotation models
Nat Commun, 15(1):6611. 2024 Aug 4.
Abstract
Identifying cellular identities is a key use case in single-cell transcriptomics. While machine learning has been leveraged to automate cell annotation predictions for some time, there has been little progress in scaling neural networks to large data sets and in constructing models that generalize well across diverse tissues. Here, we propose scTab, an automated cell type prediction model specific to tabular data, and train it using a novel data augmentation scheme across a large corpus of single-cell RNA-seq observations (22.2 million cells). In this context, we show that cross-tissue annotation requires nonlinear models and that the performance of scTab scales both in terms of training dataset size and model size. Additionally, we show that the proposed data augmentation schema improves model generalization. In summary, we introduce a de novo cell type prediction model for single-cell RNA-seq data that can be trained across a large-scale collection of curated datasets and demonstrate the benefits of using deep learning methods in this paradigm.
-
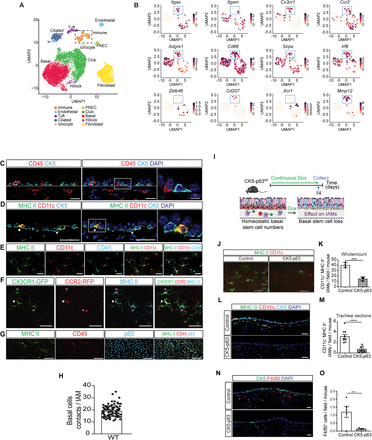
Airway basal stem cells are necessary for the maintenance of functional intraepithelial airway macrophages
bioRxiv. 2024 Jun 26.
Abstract
Adult stem cells play a crucial role in tissue homeostasis and repair through multiple mechanisms. In addition to being able to replace aged or damaged cells, stem cells provide signals that contribute to the maintenance and function of neighboring cells. In the lung, airway basal stem cells also produce cytokines and chemokines in response to inhaled irritants, allergens, and pathogens, which affect specific immune cell populations and shape the nature of the immune response. However, direct cell-to-cell signaling through contact between airway basal stem cells and immune cells has not been demonstrated. Recently, a unique population of intraepithelial airway macrophages (IAMs) has been identified in the murine trachea. Here, we demonstrate that IAMs require Notch signaling from airway basal stem cells for maintenance of their differentiated state and function. Furthermore, we demonstrate that Notch signaling between airway basal stem cells and IAMs is required for antigen-induced allergic inflammation only in the trachea where the basal stem cells are located whereas allergic responses in distal lung tissues are preserved consistent with a local circuit linking stem cells to proximate immune cells. Finally, we demonstrate that IAM-like cells are present in human conducting airways and that these cells display Notch activation, mirroring their murine counterparts. Since diverse lung stem cells have recently been identified and localized to specific anatomic niches along the proximodistal axis of the respiratory tree, we hypothesize that the direct functional coupling of local stem cell-mediated regeneration and immune responses permits a compartmentalized inflammatory response.
-
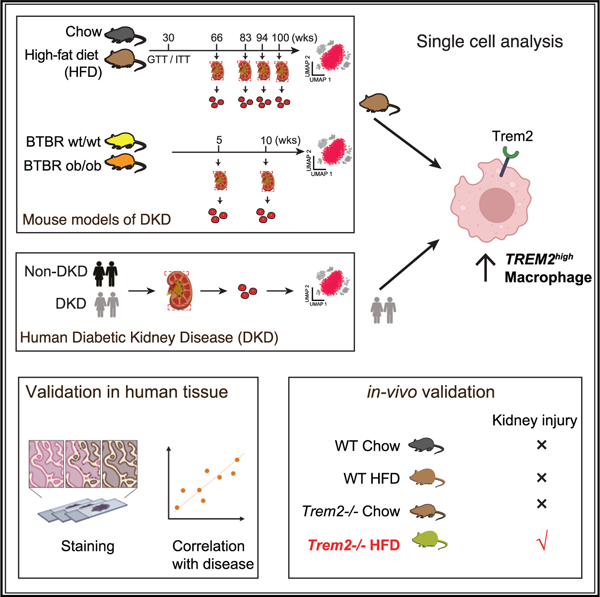
Protective role for kidney TREM2(high) macrophages in obesity- and diabetes-induced kidney injury
Cell Rep, 43(6):114253. 2024 Jun 25.
Abstract
Diabetic kidney disease (DKD), the most common cause of kidney failure, is a frequent complication of diabetes and obesity, and yet to date, treatments to halt its progression are lacking. We analyze kidney single-cell transcriptomic profiles from DKD patients and two DKD mouse models at multiple time points along disease progression-high-fat diet (HFD)-fed mice aged to 90-100 weeks and BTBR ob/ob mice (a genetic model)-and report an expanding population of macrophages with high expression of triggering receptor expressed on myeloid cells 2 (TREM2) in HFD-fed mice. TREM2(high) macrophages are enriched in obese and diabetic patients, in contrast to hypertensive patients or healthy controls in an independent validation cohort. Trem2 knockout mice on an HFD have worsening kidney filter damage and increased tubular epithelial cell injury, all signs of worsening DKD. Together, our studies suggest that strategies to enhance kidney TREM2(high) macrophages may provide therapeutic benefits for DKD.
-
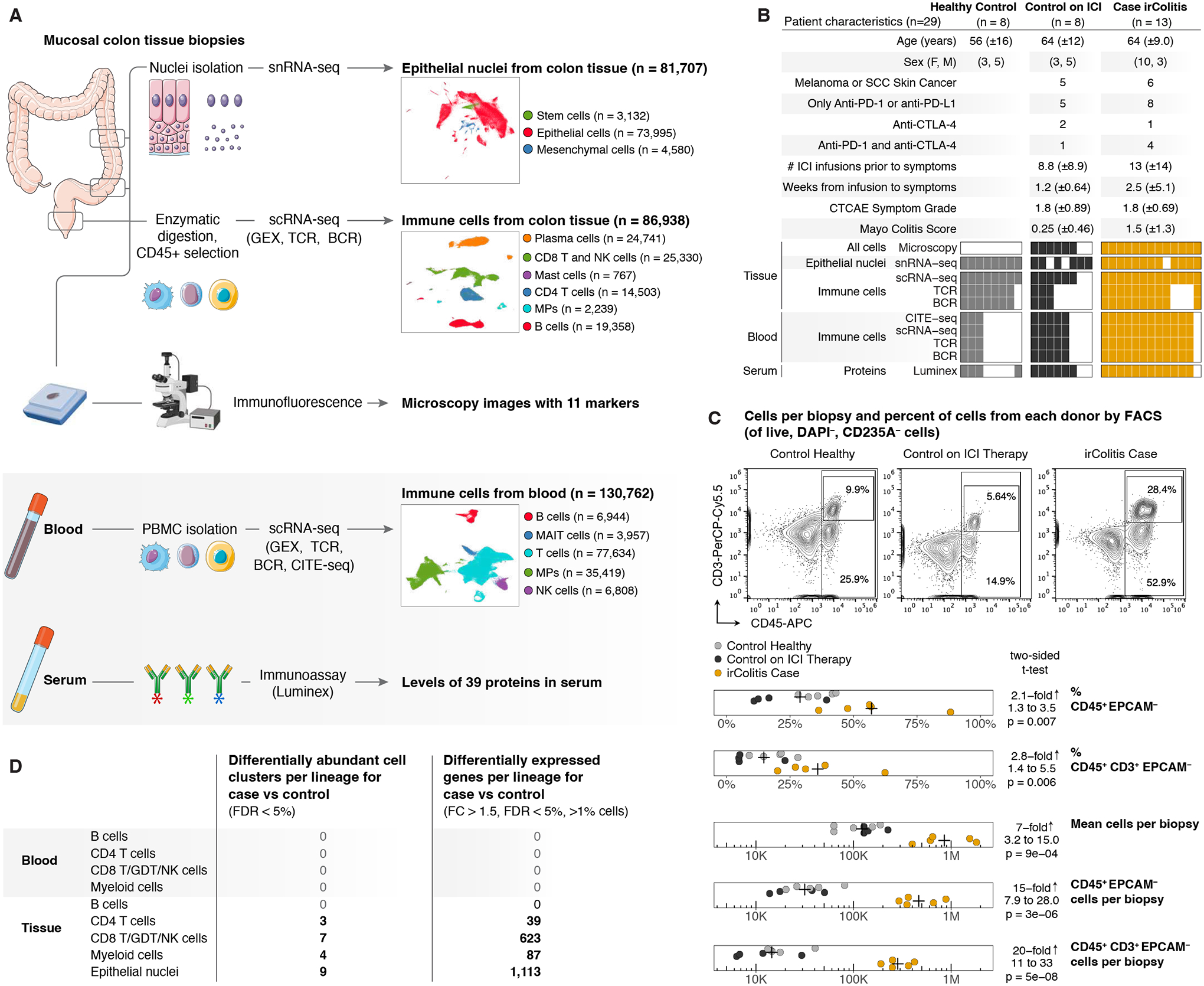
Single-cell transcriptomic analyses reveal distinct immune cell contributions to epithelial barrier dysfunction in checkpoint inhibitor colitis
Nat Med, 30(5):1349-1362. 2024 May.
Abstract
Immune checkpoint inhibitor (ICI) therapy has revolutionized oncology, but treatments are limited by immune-related adverse events, including checkpoint inhibitor colitis (irColitis). Little is understood about the pathogenic mechanisms driving irColitis, which does not readily occur in model organisms, such as mice. To define molecular drivers of irColitis, we used single-cell multi-omics to profile approximately 300,000 cells from the colon mucosa and blood of 13 patients with cancer who developed irColitis (nine on anti-PD-1 or anti-CTLA-4 monotherapy and four on dual ICI therapy; most patients had skin or lung cancer), eight controls on ICI therapy and eight healthy controls. Patients with irColitis showed expanded mucosal Tregs, ITGAE(Hi) CD8 tissue-resident memory T cells expressing CXCL13 and Th17 gene programs and recirculating ITGB2(Hi) CD8 T cells. Cytotoxic GNLY(Hi) CD4 T cells, recirculating ITGB2(Hi) CD8 T cells and endothelial cells expressing hypoxia gene programs were further expanded in colitis associated with anti-PD-1/CTLA-4 therapy compared to anti-PD-1 therapy. Luminal epithelial cells in patients with irColitis expressed PCSK9, PD-L1 and interferon-induced signatures associated with apoptosis, increased cell turnover and malabsorption. Together, these data suggest roles for circulating T cells and epithelial-immune crosstalk critical to PD-1/CTLA-4-dependent tolerance and barrier function and identify potential therapeutic targets for irColitis.
-
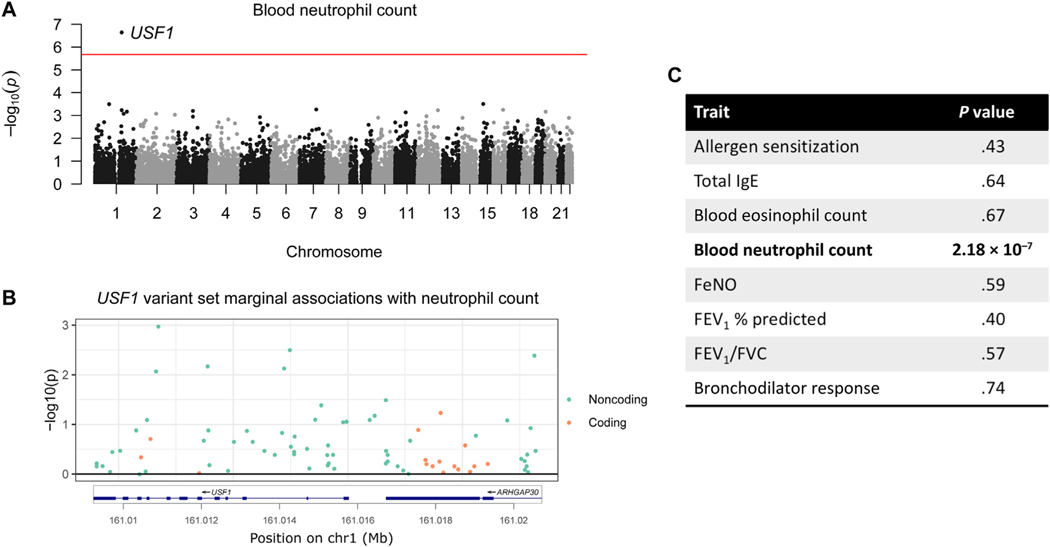
Gene-based association study of rare variants in children of diverse ancestries implicates TNFRSF21 in the development of allergic asthma
J Allergy Clin Immunol, 153(3):809-820. 2024 Mar.
Abstract
BACKGROUND: Most genetic studies of asthma and allergy have focused on common variation in individuals primarily of European ancestry. Studying the role of rare variation in quantitative phenotypes and in asthma phenotypes in populations of diverse ancestries can provide additional, important insights into the development of these traits. OBJECTIVE: We sought to examine the contribution of rare variants to different asthma- or allergy-associated quantitative traits in children with diverse ancestries and explore their role in asthma phenotypes. METHODS: We examined whole-genome sequencing data from children participants in longitudinal studies of asthma (n = 1035; parent-identified as 67% Black and 25% Hispanic) to identify rare variants (minor allele frequency < 0.01). We assigned variants to genes and tested for associations using an omnibus variant-set test between each of 24,902 genes and 8 asthma-associated quantitative traits. On combining our results with external data on predicted gene expression in humans and mouse knockout studies, we identified 3 candidate genes. A burden of rare variants in each gene and in a combined 3-gene score was tested for its associations with clinical phenotypes of asthma. Finally, published single-cell gene expression data in lower airway mucosal cells after allergen challenge were used to assess transcriptional responses to allergen. RESULTS: Rare variants in USF1 were significantly associated with blood neutrophil count (P = 2.18 × 10(-7)); rare variants in TNFRSF21 with total IgE (P = 6.47 × 10(-6)) and PIK3R6 with eosinophil count (P = 4.10 × 10(-5)) reached suggestive significance. These 3 findings were supported by independent data from human and mouse studies. A burden of rare variants in TNFRSF21 and in a 3-gene score was associated with allergy-related phenotypes in cohorts of children with mild and severe asthma. Furthermore, TNFRSF21 was significantly upregulated in bronchial basal epithelial cells from adults with allergic asthma but not in adults with allergies (but not asthma) after allergen challenge. CONCLUSIONS: We report novel associations between rare variants in genes and allergic and inflammatory phenotypes in children with diverse ancestries, highlighting TNFRSF21 as contributing to the development of allergic asthma.
-
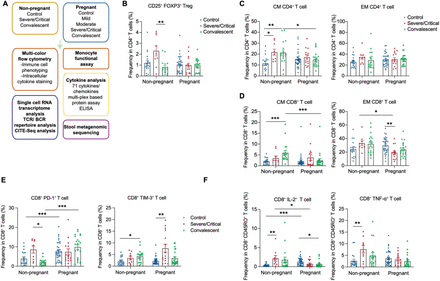
SARS-CoV-2 infection elucidates unique features of pregnancy-specific immunity
medRxiv. 2024 Feb 7.
Abstract
Pregnancy is a risk factor for increased severity of SARS-CoV-2 and other respiratory infections. The mechanisms underlying this risk have not been well-established, partly due to a limited understanding of how pregnancy shapes immune responses. To gain insight into the role of pregnancy in modulating immune responses at steady state and upon perturbation, we collected peripheral blood mononuclear cells (PBMC), plasma, and stool from 226 women, including 152 pregnant individuals (n = 96 with SARS-CoV-2 infection and n = 56 healthy controls) and 74 non-pregnant women (n = 55 with SARS-CoV-2 and n = 19 healthy controls). We found that SARS-CoV-2 infection was associated with altered T cell responses in pregnant compared to non-pregnant women. Differences included a lower percentage of memory T cells, a distinct clonal expansion of CD4-expressing CD8 (+) T cells, and the enhanced expression of T cell exhaustion markers, such as programmed cell death-1 (PD-1) and T cell immunoglobulin and mucin domain-3 (Tim-3), in pregnant women. We identified additional evidence of immune dysfunction in severely and critically ill pregnant women, including a lack of expected elevation in regulatory T cell (Treg) levels, diminished interferon responses, and profound suppression of monocyte function. Consistent with earlier data, we found maternal obesity was also associated with altered immune responses to SARS-CoV-2 infection, including enhanced production of inflammatory cytokines by T cells. Certain gut bacterial species were altered in pregnancy and upon SARS-CoV-2 infection in pregnant individuals compared to non-pregnant women. Shifts in cytokine and chemokine levels were also identified in the sera of pregnant individuals, most notably a robust increase of interleukin-27 (IL-27), a cytokine known to drive T cell exhaustion, in the pregnant uninfected control group compared to all non-pregnant groups. IL-27 levels were also significantly higher in uninfected pregnant controls compared to pregnant SARS-CoV-2-infected individuals. Using two different preclinical mouse models of inflammation-induced fetal demise and respiratory influenza viral infection, we found that enhanced IL-27 protects developing fetuses from maternal inflammation but renders adult female mice vulnerable to viral infection. These combined findings from human and murine studies reveal nuanced pregnancy-associated immune responses, suggesting mechanisms underlying the increased susceptibility of pregnant individuals to viral respiratory infections.
2023 (8)
-
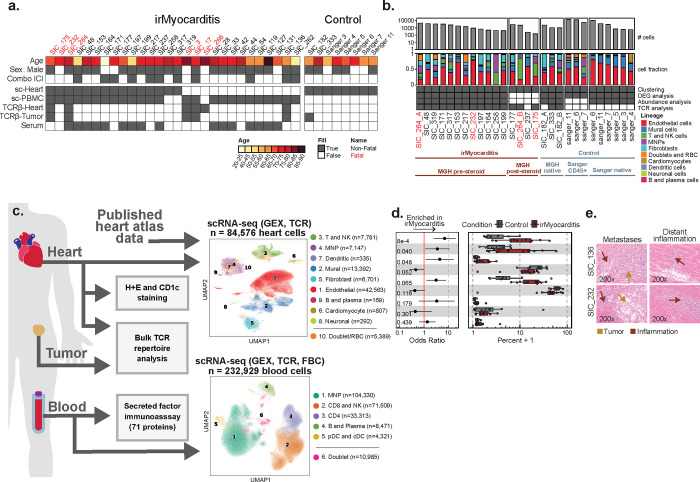
Immune Responses in Checkpoint Myocarditis Across Heart, Blood, and Tumor
bioRxiv. 2023 Nov 29.
Abstract
Immune checkpoint inhibitors (ICIs) are widely used anti-cancer therapies that can cause morbid and potentially fatal immune-related adverse events (irAEs). ICI-related myocarditis (irMyocarditis) is uncommon but has the highest mortality of any irAE. The pathogenesis of irMyocarditis and its relationship to anti-tumor immunity remain poorly understood. We sought to define immune responses in heart, tumor, and blood during irMyocarditis and identify biomarkers of clinical severity by leveraging single-cell (sc)RNA-seq coupled with T cell receptor (TCR) sequencing, microscopy, and proteomics analysis of 28 irMyocarditis patients and 23 controls. Our analysis of 284,360 cells from heart and blood specimens identified cytotoxic T cells, inflammatory macrophages, conventional dendritic cells (cDCs), and fibroblasts enriched in irMyocarditis heart tissue. Additionally, potentially targetable, pro-inflammatory transcriptional programs were upregulated across multiple cell types. TCR clones enriched in heart and paired tumor tissue were largely non-overlapping, suggesting distinct T cell responses within these tissues. We also identify the presence of cardiac-expanded TCRs in a circulating, cycling CD8 T cell population as a novel peripheral biomarker of fatality. Collectively, these findings highlight critical biology driving irMyocarditis and putative biomarkers for therapeutic intervention.
-

Single-cell RNA sequencing of murine ankle joints over time reveals distinct transcriptional changes following Borrelia burgdorferi infection
iScience, 26(11):108217. 2023 Nov 17.
Abstract
Lyme disease is caused by the bacterial pathogen Borrelia burgdorferi, which can be readily modeled in laboratory mice. In order to understand the cellular and transcriptional changes that occur during B. burgdorferi infection, we conducted single-cell RNA sequencing (scRNA-seq) of ankle joints of infected C57BL/6 mice over time. We found that macrophages/monocytes, T cells, synoviocytes and fibroblasts all showed significant differences in gene expression of both inflammatory and non-inflammatory genes that peaked early and returned to baseline before the typical resolution of arthritis. Predictions of cellular interactions showed that macrophages appear to communicate extensively between different clusters of macrophages as well as with fibroblasts and synoviocytes. Our data give unique insights into the interactions between B. burgdorferi and the murine immune system over time and allow for a better understanding of mechanisms by which the dysregulation of the immune response may lead to prolonged symptoms in some patients.
-
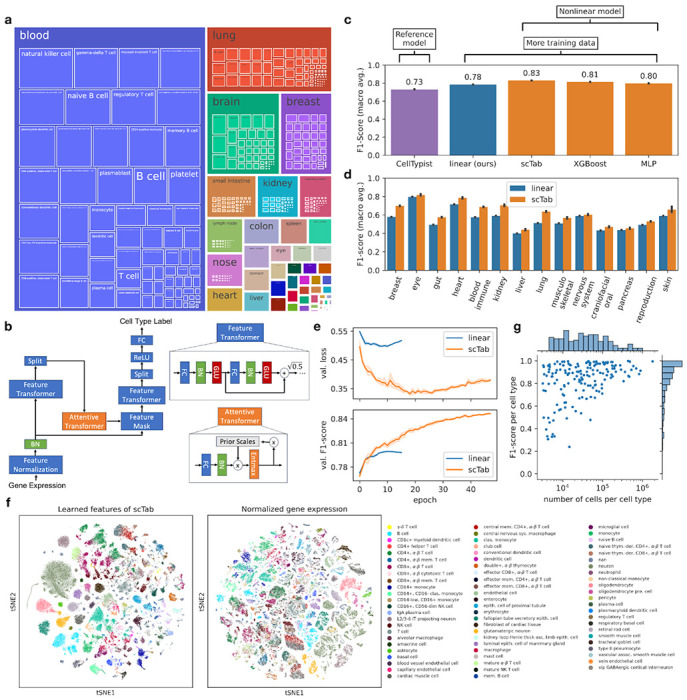
Scaling cross-tissue single-cell annotation models
bioRxiv. 2023 Oct 10.
Abstract
Identifying cellular identities (both novel and well-studied) is one of the key use cases in single-cell transcriptomics. While supervised machine learning has been leveraged to automate cell annotation predictions for some time, there has been relatively little progress both in scaling neural networks to large data sets and in constructing models that generalize well across diverse tissues and biological contexts up to whole organisms. Here, we propose scTab, an automated, feature-attention-based cell type prediction model specific to tabular data, and train it using a novel data augmentation scheme across a large corpus of single-cell RNA-seq observations (22.2 million human cells in total). In addition, scTab leverages deep ensembles for uncertainty quantification. Moreover, we account for ontological relationships between labels in the model evaluation to accommodate for differences in annotation granularity across datasets. On this large-scale corpus, we show that cross-tissue annotation requires nonlinear models and that the performance of scTab scales in terms of training dataset size as well as model size - demonstrating the advantage of scTab over current state-of-the-art linear models in this context. Additionally, we show that the proposed data augmentation schema improves model generalization. In summary, we introduce a de novo cell type prediction model for single-cell RNA-seq data that can be trained across a large-scale collection of curated datasets from a diverse selection of human tissues and demonstrate the benefits of using deep learning methods in this paradigm. Our codebase, training data, and model checkpoints are publicly available at https://github.com/theislab/scTab to further enable rigorous benchmarks of foundation models for single-cell RNA-seq data.
-
Resident memory T cell development is associated with AP-1 transcription factor upregulation across anatomical niches
bioRxiv. 2023 Oct 2.
Abstract
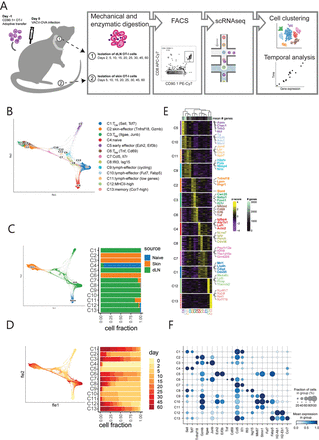
Tissue-resident memory T (T (RM) ) cells play a central role in immune responses to pathogens across all barrier tissues after infection. However, the underlying mechanisms that drive T (RM) differentiation and priming for their recall effector function remains unclear. In this study, we leveraged both newly generated and publicly available single-cell RNA-sequencing (scRNAseq) data generated across 10 developmental time points to define features of CD8 T (RM) across both skin and small-intestine intraepithelial lymphocytes (siIEL). We employed linear modeling to capture temporally-associated gene programs that increase their expression levels in T cell subsets transitioning from an effector to a memory T cell state. In addition to capturing tissue-specific gene programs, we defined a consensus T (RM) signature of 60 genes across skin and siIEL that can effectively distinguish T (RM) from circulating T cell populations, providing a more specific T (RM) signature than what was previously generated by comparing bulk T (RM) to naïve or non-tissue resident memory populations. This updated T (RM) signature included the AP-1 transcription factor family members Fos, Fosb and Fosl2 . Moreover, ATACseq analysis detected an enrichment of AP-1-specific motifs at open chromatin sites in mature T (RM) . CyCIF tissue imaging detected nuclear co-localization of AP-1 members Fosb and Junb in resting CD8 T (RM) >100 days post-infection. Taken together, these results reveal a critical role of AP-1 transcription factor members in T (RM) biology and suggests a novel mechanism for rapid reactivation of resting T (RM) in tissue upon antigen encounter.
-
The evolving landscape of immune-related adverse events that follow immune checkpoint immunotherapy in cancer patients
Immunol Rev, 318(1):4-10. 2023 Sep.
-
A human model of asthma exacerbation reveals transcriptional programs and cell circuits specific to allergic asthma
Sci Immunol, 8(83):eabq6352. 2023 May 12.
Abstract
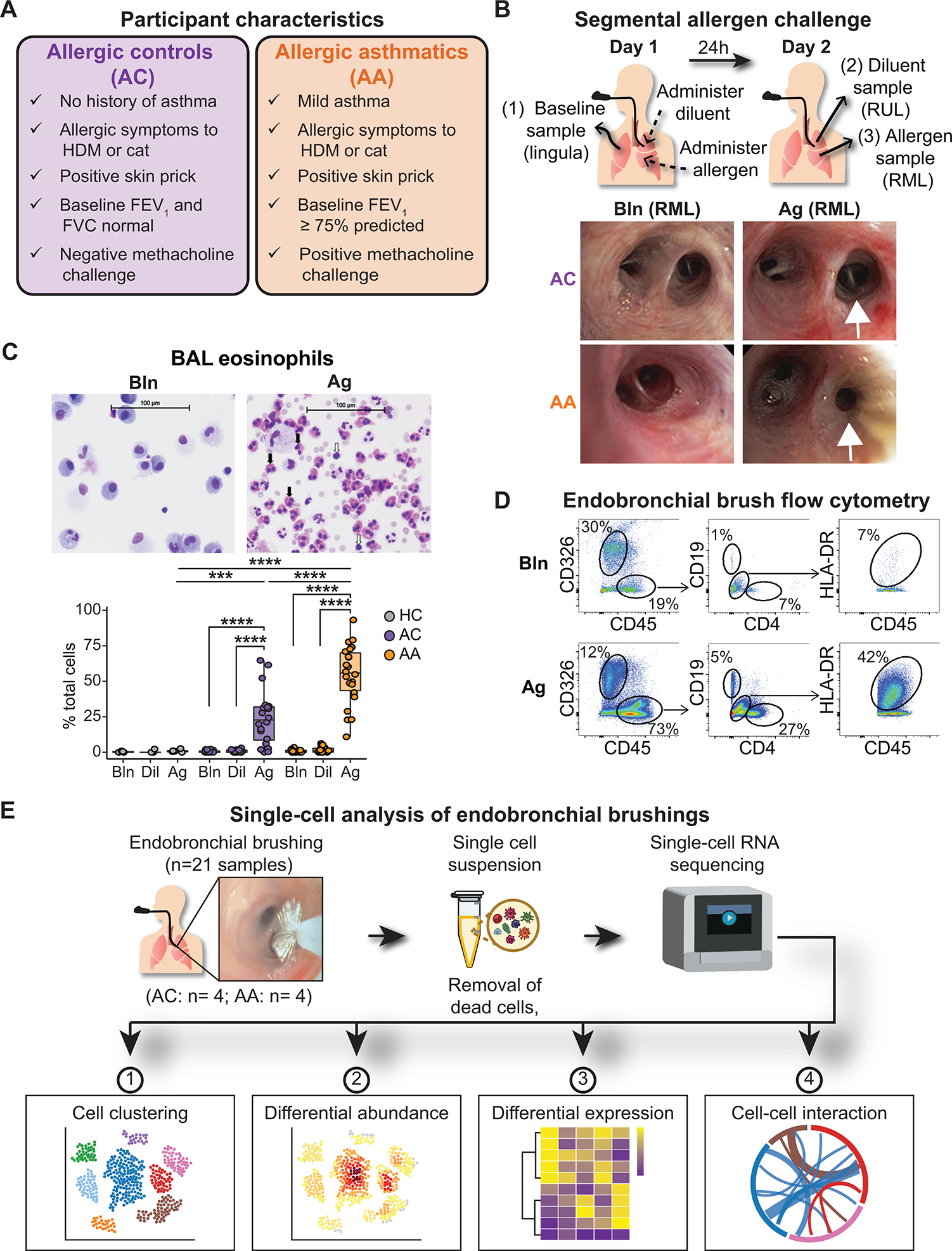
Asthma is a chronic disease most commonly associated with allergy and type 2 inflammation. However, the mechanisms that link airway inflammation to the structural changes that define asthma are incompletely understood. Using a human model of allergen-induced asthma exacerbation, we compared the lower airway mucosa in allergic asthmatics and allergic non-asthmatic controls using single-cell RNA sequencing. In response to allergen, the asthmatic airway epithelium was highly dynamic and up-regulated genes involved in matrix degradation, mucus metaplasia, and glycolysis while failing to induce injury-repair and antioxidant pathways observed in controls. IL9-expressing pathogenic T(H)2 cells were specific to asthmatic airways and were only observed after allergen challenge. Additionally, conventional type 2 dendritic cells (DC2 that express CD1C) and CCR2-expressing monocyte-derived cells (MCs) were uniquely enriched in asthmatics after allergen, with up-regulation of genes that sustain type 2 inflammation and promote pathologic airway remodeling. In contrast, allergic controls were enriched for macrophage-like MCs that up-regulated tissue repair programs after allergen challenge, suggesting that these populations may protect against asthmatic airway remodeling. Cellular interaction analyses revealed a T(H)2-mononuclear phagocyte-basal cell interactome unique to asthmatics. These pathogenic cellular circuits were characterized by type 2 programming of immune and structural cells and additional pathways that may sustain and amplify type 2 signals, including TNF family signaling, altered cellular metabolism, failure to engage antioxidant responses, and loss of growth factor signaling. Our findings therefore suggest that pathogenic effector circuits and the absence of proresolution programs drive structural airway disease in response to type 2 inflammation.
-
Protocol for bulk RNA sequencing of enriched human neutrophils from whole blood and estimation of sample purity
STAR Protoc, 4(1):102125. 2023 Mar 17.
Abstract
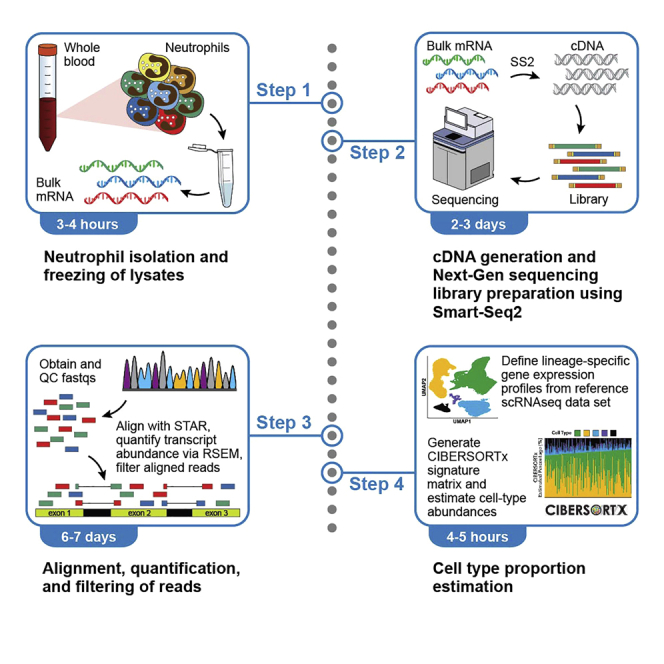
Although neutrophils are the most abundant leukocyte in healthy individuals and impact outcomes of diseases ranging from sepsis to cancer, they remain understudied due to technical constraints of isolation, preservation, and sequencing. We present a modified Smart-Seq2 protocol for bulk RNA sequencing of neutrophils enriched from whole blood. We describe steps for neutrophil isolation, cDNA generation, library preparation, and sample purity estimation via a bioinformatic approach. Our approach permits the collection of large cohorts and enables detection of neutrophil transcriptomic subtypes. For complete details on the use and execution of this protocol, please refer to LaSalle et al. (2022)(1) and Boribong et al. (2022).(2).
-
Soluble and cell-based markers of immune checkpoint inhibitor-associated nephritis
J Immunother Cancer, 11(1). 2023 Jan.
Abstract
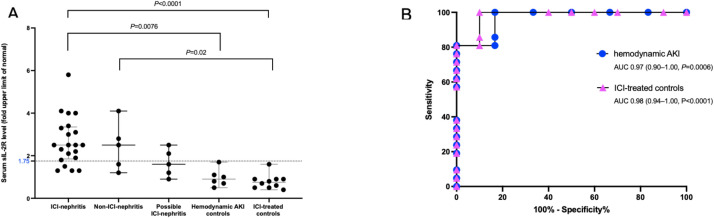
BACKGROUND: Non-invasive biomarkers of immune checkpoint inhibitor-associated acute tubulointerstitial nephritis (ICI-nephritis) are urgently needed. Because ICIs block immune checkpoint pathways that include cytotoxic T lymphocyte antigen 4 (CTLA4), we hypothesized that biomarkers of immune dysregulationpreviously defined in patients with congenital CTLA4 deficiency, including elevated soluble interleukin-2 receptor alpha (sIL-2R) and flow cytometric cell-based markers of B and T cell dysregulation in peripheral blood may aid the diagnosis of ICI-nephritis. METHODS: A retrospective cohort of patients diagnosed with ICI-nephritis was compared with three prospectively enrolled control cohorts: ICI-treated controls without immune-related adverse events, patients not on ICIs with hemodynamic acute kidney injury (hemodynamic AKI), and patients not on ICIs with biopsy proven acute interstitial nephritis from other causes (non-ICI-nephritis). sIL-2R level and flow cytometric parameters were compared between groups using Wilcoxon rank sum test or Kruskal-Wallis test. Receiver operating characteristic curves were generated to define the accuracy of sIL-2R and flow cytometric biomarkers in diagnosing ICI-nephritis. The downstream impact of T cell activation in the affected kidney was investigated using archived biopsy samples to evaluate the gene expression of IL2RA, IL-2 signaling, and T cell receptor signaling in patients with ICI-nephritis compared with other causes of drug-induced nephritis, acute tubular injury, and histologically normal controls. RESULTS: sIL-2R level in peripheral blood was significantly higher in patients with ICI-nephritis (N=24) (median 2.5-fold upper limit of normal (ULN), IQR 1.9-3.3), compared with ICI-treated controls (N=10) (median 0.8-fold ULN, IQR 0.5-0.9, p<0.001) and hemodynamic AKI controls (N=6) (median 0.9-fold-ULN, IQR 0.7-1.1, p=0.008). A sIL-2R cut-off point of 1.75-fold ULN was highly diagnostic of ICI-nephritis (area under the curve >96%) when compared with either ICI-treated or hemodynamic AKI controls. By peripheral blood flow cytometry analysis, lower absolute CD8+T cells, CD45RA+CD8+ T cells, memory CD27+B cells, and expansion of plasmablasts were prominent features of ICI-nephritis compared with ICI-treated controls. Gene expressions for IL2RA, IL-2 signaling, and T cell receptor signaling in the kidney tissue with ICI-nephritis were significantly higher compared with controls. CONCLUSION: Elevated sIL-2R level and flow cytometric markers of both B and T cell dysregulation may aid the diagnosis of ICI-nephritis.
2022 (8)
-

Germline variants associated with toxicity to immune checkpoint blockade
Nat Med, 28(12):2584-2591. 2022 Dec.
Abstract
Immune checkpoint inhibitors (ICIs) have yielded remarkable responses but often lead to immune-related adverse events (irAEs). Although germline causes for irAEs have been hypothesized, no individual variant associated with developing irAEs has been identified. We carried out a genome-wide association study of 1,751 patients on ICIs across 12 cancer types. We investigated two irAE phenotypes: (1) high-grade (3-5) and (2) all-grade events. We identified 3 genome-wide significant associations (P < 5 × 10(-8)) in the discovery cohort associated with all-grade irAEs: rs16906115 near IL7 (combined P = 3.6 × 10(-11); hazard ratio (HR) = 2.1); rs75824728 near IL22RA1 (combined P = 3.5 × 10(-8); HR = 1.8); and rs113861051 on 4p15 (combined P = 1.2 × 10(-8), HR = 2.0); rs16906115 was replicated in 3 independent studies. The association near IL7 colocalized with the gain of a new cryptic exon for IL7, a critical regulator of lymphocyte homeostasis. Patients carrying the IL7 germline variant exhibited significantly increased lymphocyte stability after ICI initiation, which was itself predictive of downstream irAEs and improved survival.
-
-
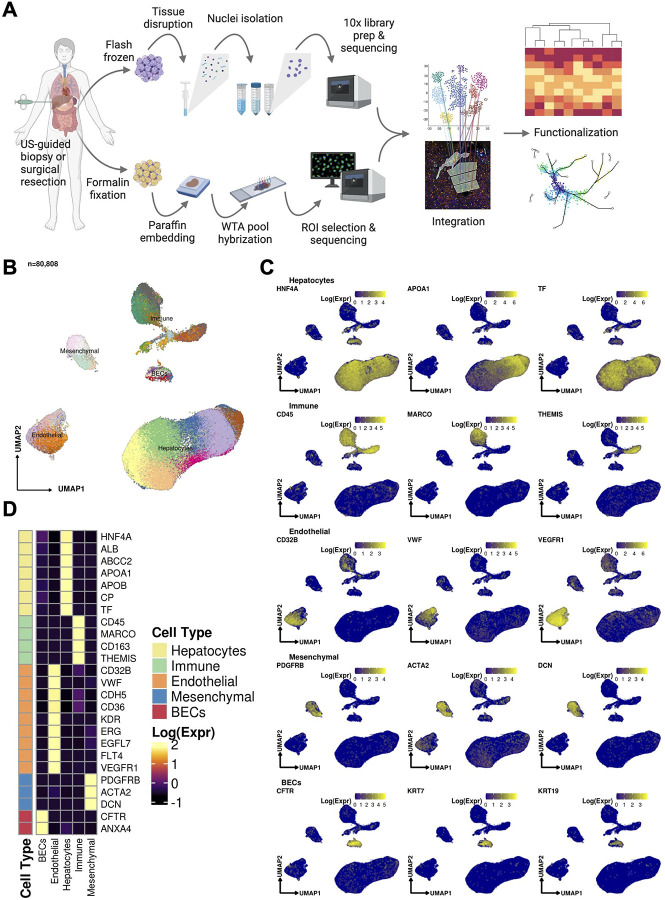
A single-nucleus and spatial transcriptomic atlas of the COVID-19 liver reveals topological, functional, and regenerative organ disruption in patients
bioRxiv. 2022 Oct 28.
Abstract
The molecular underpinnings of organ dysfunction in acute COVID-19 and its potential long-term sequelae are under intense investigation. To shed light on these in the context of liver function, we performed single-nucleus RNA-seq and spatial transcriptomic profiling of livers from 17 COVID-19 decedents. We identified hepatocytes positive for SARS-CoV-2 RNA with an expression phenotype resembling infected lung epithelial cells. Integrated analysis and comparisons with healthy controls revealed extensive changes in the cellular composition and expression states in COVID-19 liver, reflecting hepatocellular injury, ductular reaction, pathologic vascular expansion, and fibrogenesis. We also observed Kupffer cell proliferation and erythrocyte progenitors for the first time in a human liver single-cell atlas, resembling similar responses in liver injury in mice and in sepsis, respectively. Despite the absence of a clinical acute liver injury phenotype, endothelial cell composition was dramatically impacted in COVID-19, concomitantly with extensive alterations and profibrogenic activation of reactive cholangiocytes and mesenchymal cells. Our atlas provides novel insights into liver physiology and pathology in COVID-19 and forms a foundational resource for its investigation and understanding.
-
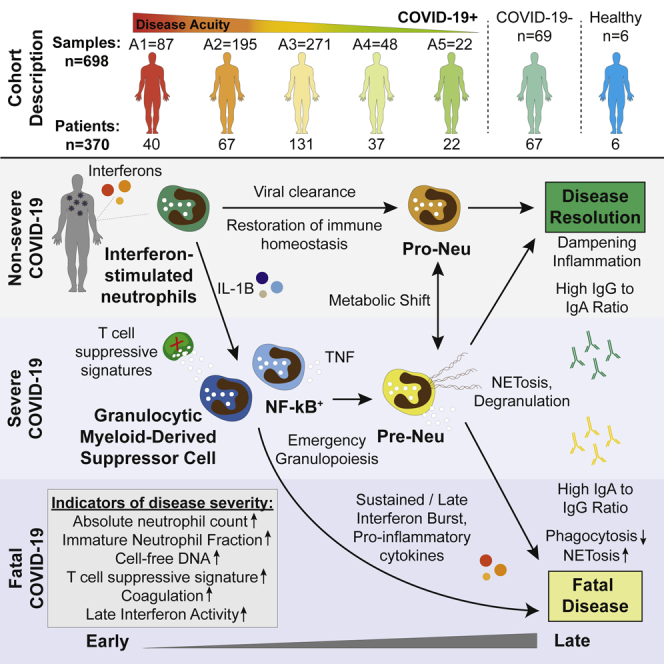
Longitudinal characterization of circulating neutrophils uncovers phenotypes associated with severity in hospitalized COVID-19 patients
Cell Rep Med, 3(10):100779. 2022 Oct 18.
Abstract
Mechanisms of neutrophil involvement in severe coronavirus disease 2019 (COVID-19) remain incompletely understood. Here, we collect longitudinal blood samples from 306 hospitalized COVID-19(+) patients and 86 controls and perform bulk RNA sequencing of enriched neutrophils, plasma proteomics, and high-throughput antibody profiling to investigate relationships between neutrophil states and disease severity. We identify dynamic switches between six distinct neutrophil subtypes. At days 3 and 7 post-hospitalization, patients with severe disease display a granulocytic myeloid-derived suppressor cell-like gene expression signature, while patients with resolving disease show a neutrophil progenitor-like signature. Humoral responses are identified as potential drivers of neutrophil effector functions, with elevated severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-specific immunoglobulin G1 (IgG1)-to-IgA1 ratios in plasma of severe patients who survived. In vitro experiments confirm that while patient-derived IgG antibodies induce phagocytosis in healthy donor neutrophils, IgA antibodies predominantly induce neutrophil cell death. Overall, our study demonstrates a dysregulated myelopoietic response in severe COVID-19 and a potential role for IgA-dominant responses contributing to mortality.
-
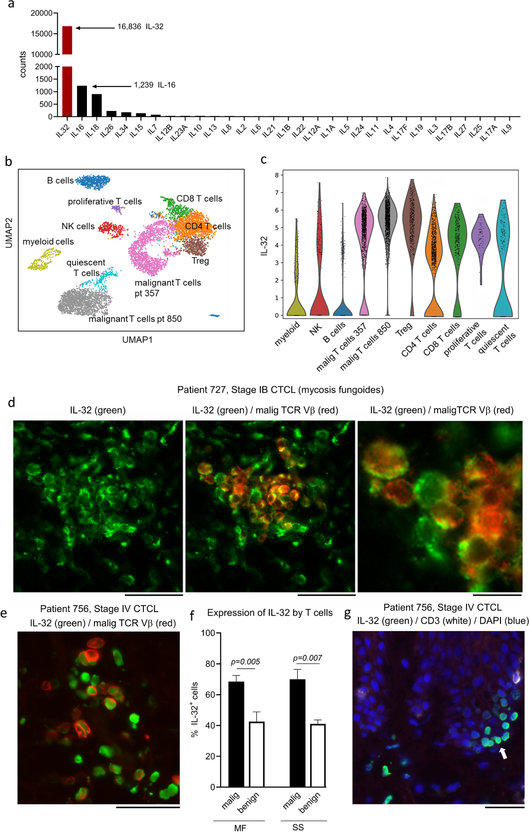
-
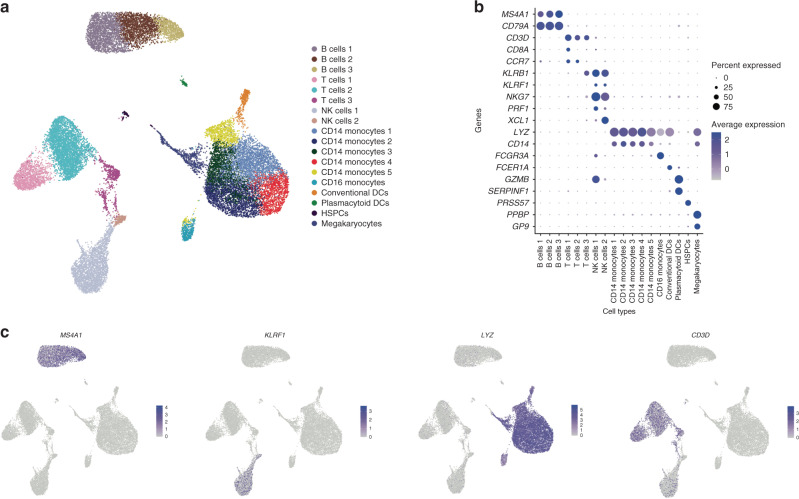
Single-cell immunophenotyping of the fetal immune response to maternal SARS-CoV-2 infection in late gestation
Pediatr Res, 91(5):1090-1098. 2022 Apr.
Abstract
BACKGROUND: During the COVID-19 pandemic, thousands of pregnant women have been infected with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The implications of maternal SARS-CoV-2 infection on fetal and childhood well-being need to be characterized. We aimed to characterize the fetal immune response to maternal SARS-CoV-2 infection. METHODS: We performed single-cell RNA-sequencing and T cell receptor sequencing on cord blood mononuclear cells (CBMCs) from newborns of mothers infected with SARS-CoV-2 in the third trimester (cases) or without SARS-CoV-2 infection (controls). RESULTS: We identified widespread gene expression changes in CBMCs from cases, including upregulation of interferon-stimulated genes and major histocompatibility complex genes in CD14(+) monocytes, transcriptional changes suggestive of activation of plasmacytoid dendritic cells, and activation and exhaustion of natural killer cells. Lastly, we observed fetal T cell clonal expansion in cases compared to controls. CONCLUSIONS: As none of the infants were infected with SARS-CoV-2, our results suggest that maternal SARS-CoV-2 infection might modulate the fetal immune system in the absence of vertical transmission. IMPACT: The implications of maternal SARS-CoV-2 infection in the absence of vertical transmission on fetal and childhood well-being are poorly understood. Maternal SARS-CoV-2 infection might modulate the fetal immune system in the absence of vertical transmission. This study raises important questions about the untoward effects of maternal SARS-CoV-2 on the fetus, even in the absence of vertical transmission.
-
Plasmacytoid dendritic cells: Welcome back to the DC fold
Immunity, 55(3):380-382. 2022 Mar 8.
Abstract
The presumed common origin of plasmacytoid and conventional dendritic cells has been the contentious subject of recent debate. In this issue of Immunity, Feng et al. employed an inducible cell barcoding system to track clonal relationships and uncovered a surprising close developmental relationship between cDC1s and pDCs.
-
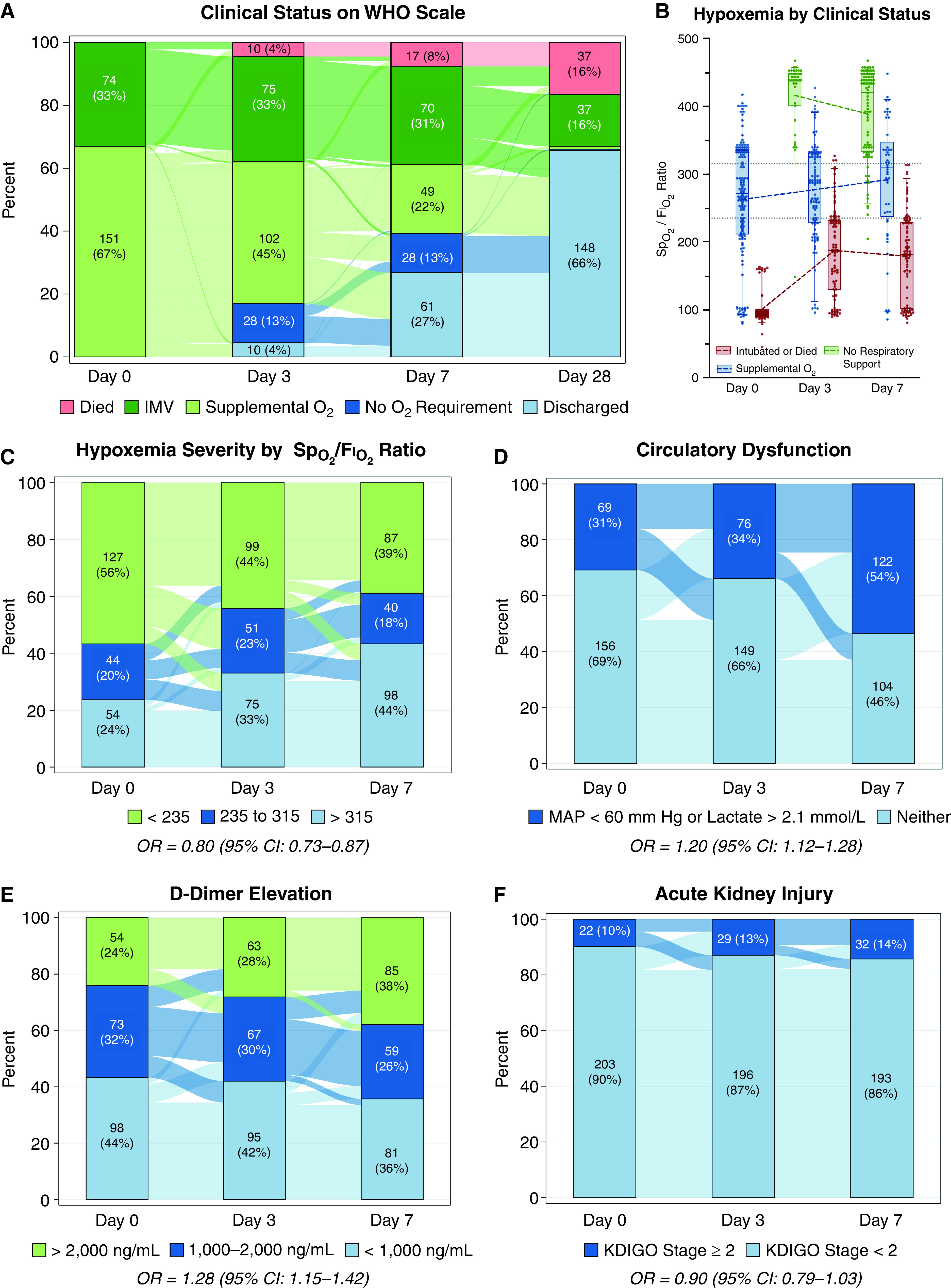
Alveolar, Endothelial, and Organ Injury Marker Dynamics in Severe COVID-19
Am J Respir Crit Care Med, 205(5):507-519. 2022 Mar 1.
Abstract
Rationale: Alveolar and endothelial injury may be differentially associated with coronavirus disease (COVID-19) severity over time. Objectives: To describe alveolar and endothelial injury dynamics and associations with COVID-19 severity, cardiorenovascular injury, and outcomes. Methods: This single-center observational study enrolled patients with COVID-19 requiring respiratory support at emergency department presentation. More than 40 markers of alveolar (including receptor for advanced glycation endproducts [RAGE]), endothelial (including angiopoietin-2), and cardiorenovascular injury (including renin, kidney injury molecule-1, and troponin-I) were serially compared between invasively and spontaneously ventilated patients using mixed-effects repeated-measures models. Ventilatory ratios were calculated for intubated patients. Associations of biomarkers with modified World Health Organization scale at Day 28 were determined with multivariable proportional-odds regression. Measurements and Main Results: Of 225 patients, 74 (33%) received invasive ventilation at Day 0. RAGE was 1.80-fold higher in invasive ventilation patients at Day 0 (95% confidence interval [CI], 1.50-2.17) versus spontaneous ventilation, but decreased over time in all patients. Changes in alveolar markers did not correlate with changes in endothelial, cardiac, or renal injury markers. In contrast, endothelial markers were similar to lower at Day 0 for invasive ventilation versus spontaneous ventilation, but then increased over time only among intubated patients. In intubated patients, angiopoietin-2 was similar (fold difference, 1.02; 95% CI, 0.89-1.17) to nonintubated patients at Day 0 but 1.80-fold higher (95% CI, 1.56-2.06) at Day 3; cardiorenovascular injury markers showed similar patterns. Endothelial markers were not consistently associated with ventilatory ratios. Endothelial markers were more often significantly associated with 28-day outcomes than alveolar markers. Conclusions: Alveolar injury markers increase early. Endothelial injury markers increase later and are associated with cardiorenovascular injury and 28-day outcome. Alveolar and endothelial injury likely contribute at different times to disease progression in severe COVID-19.
2021 (18)
-

Harnessing the Potential of Multiomics Studies for Precision Medicine in Infectious Disease
Open Forum Infect Dis, 8(11):ofab483. 2021 Nov.
Abstract
The field of infectious diseases currently takes a reactive approach and treats infections as they present in patients. Although certain populations are known to be at greater risk of developing infection (eg, immunocompromised), we lack a systems approach to define the true risk of future infection for a patient. Guided by impressive gains in "omics" technologies, future strategies to infectious diseases should take a precision approach to infection through identification of patients at intermediate and high-risk of infection and deploy targeted preventative measures (ie, prophylaxis). The advances of high-throughput immune profiling by multiomics approaches (ie, transcriptomics, epigenomics, metabolomics, proteomics) hold the promise to identify patients at increased risk of infection and enable risk-stratifying approaches to be applied in the clinic. Integration of patient-specific data using machine learning improves the effectiveness of prediction, providing the necessary technologies needed to propel the field of infectious diseases medicine into the era of personalized medicine.
-
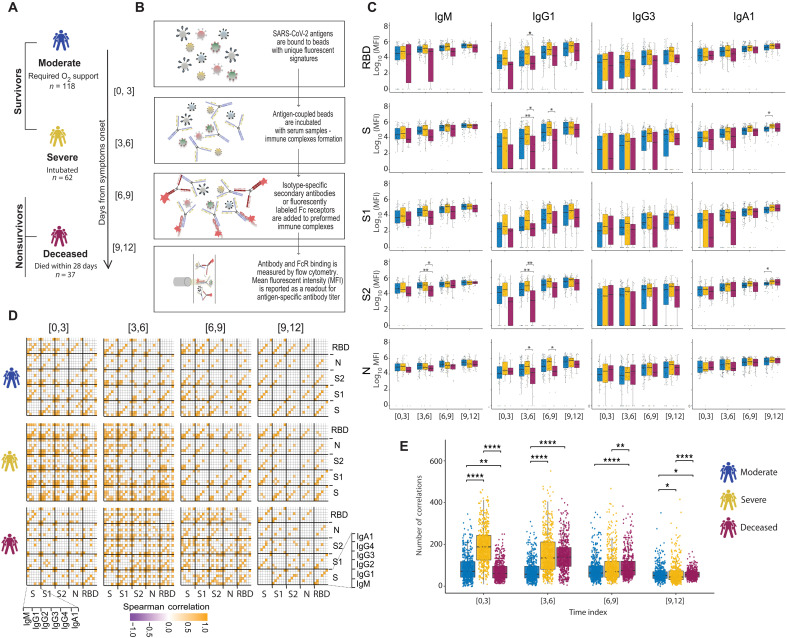
Early cross-coronavirus reactive signatures of humoral immunity against COVID-19
Sci Immunol, 6(64):eabj2901. 2021 Oct 15.
Abstract
The introduction of vaccines has inspired hope in the battle against SARS-CoV-2. However, the emergence of viral variants, in the absence of potent antivirals, has left the world struggling with the uncertain nature of this disease. Antibodies currently represent the strongest correlate of immunity against SARS-CoV-2, thus we profiled the earliest humoral signatures in a large cohort of acutely ill (survivors and nonsurvivors) and mild or asymptomatic individuals with COVID-19. Although a SARS-CoV-2–specific immune response evolved rapidly in survivors of COVID-19, nonsurvivors exhibited blunted and delayed humoral immune evolution, particularly with respect to S2-specific antibodies. Given the conservation of S2 across β-coronaviruses, we found that the early development of SARS-CoV-2–specific immunity occurred in tandem with preexisting common β-coronavirus OC43 humoral immunity in survivors, which was also selectively expanded in individuals that develop a paucisymptomatic infection. These data point to the importance of cross-coronavirus immunity as a correlate of protection against COVID-19.
-
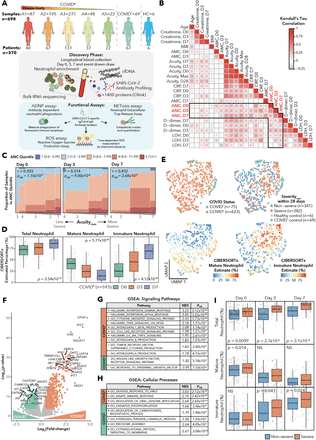
Longitudinal characterization of circulating neutrophils uncovers distinct phenotypes associated with disease severity in hospitalized COVID-19 patients
bioRxiv. 2021 Oct 5.
Abstract
Multiple studies have identified an association between neutrophils and COVID-19 disease severity; however, the mechanistic basis of this association remains incompletely understood. Here we collected 781 longitudinal blood samples from 306 hospitalized COVID-19 (+) patients, 78 COVID-19 (âˆ') acute respiratory distress syndrome patients, and 8 healthy controls, and performed bulk RNA-sequencing of enriched neutrophils, plasma proteomics, cfDNA measurements and high throughput antibody profiling assays to investigate the relationship between neutrophil states and disease severity or death. We identified dynamic switches between six distinct neutrophil subtypes using non-negative matrix factorization (NMF) clustering. At days 3 and 7 post-hospitalization, patients with severe disease had an enrichment of a granulocytic myeloid derived suppressor cell-like state gene expression signature, while non-severe patients with resolved disease were enriched for a progenitor-like immature neutrophil state signature. Severe disease was associated with gene sets related to neutrophil degranulation, neutrophil extracellular trap (NET) signatures, distinct metabolic signatures, and enhanced neutrophil activation and generation of reactive oxygen species (ROS). We found that the majority of patients had a transient interferon-stimulated gene signature upon presentation to the emergency department (ED) defined here as Day 0, regardless of disease severity, which persisted only in patients who subsequently died. Humoral responses were identified as potential drivers of neutrophil effector functions, as enhanced antibody-dependent neutrophil phagocytosis and reduced NETosis was associated with elevated SARS-CoV-2-specific IgG1-to-IgA1 ratios in plasma of severe patients who survived. In vitro experiments confirmed that while patient-derived IgG antibodies mostly drove neutrophil phagocytosis and ROS production in healthy donor neutrophils, patient-derived IgA antibodies induced a predominant NETosis response. Overall, our study demonstrates neutrophil dysregulation in severe COVID-19 and a potential role for IgA-dominant responses in driving neutrophil effector functions in severe disease and mortality.
-
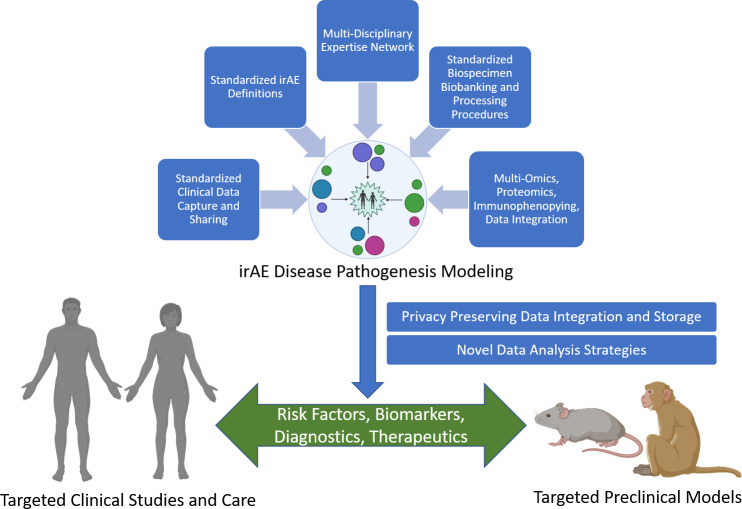
Development of preclinical and clinical models for immune-related adverse events following checkpoint immunotherapy: a perspective from SITC and AACR
J Immunother Cancer, 9(9). 2021 Sep.
Abstract
Recent advances in cancer immunotherapy have completely revolutionized cancer treatment strategies. Nonetheless, the increasing incidence of immune-related adverse events (irAEs) is now limiting the overall benefits of these treatments. irAEs are well-recognized side effects of some of the most effective cancer immunotherapy agents, including antibody blockade of the cytotoxic T-lymphocyte-associated protein 4 and programmed death protein 1/programmed-death ligand 1 pathways. To develop an action plan on the key elements needed to unravel and understand the key mechanisms driving irAEs, the Society for Immunotherapy for Cancer and the American Association for Cancer Research partnered to bring together research and clinical experts in cancer immunotherapy, autoimmunity, immune regulation, genetics and informatics who are investigating irAEs using animal models, clinical data and patient specimens to discuss current strategies and identify the critical next steps needed to create breakthroughs in our understanding of these toxicities. The genetic and environmental risk factors, immune cell subsets and other key immunological mediators and the unique clinical presentations of irAEs across the different organ systems were the foundation for identifying key opportunities and future directions described in this report. These include the pressing need for significantly improved preclinical model systems, broader collection of biospecimens with standardized collection and clinical annotation made available for research and integration of electronic health record and multiomic data with harmonized and standardized methods, definitions and terminologies to further our understanding of irAE pathogenesis. Based on these needs, this report makes a set of recommendations to advance our understanding of irAE mechanisms, which will be crucial to prevent their occurrence and improve their treatment.
-
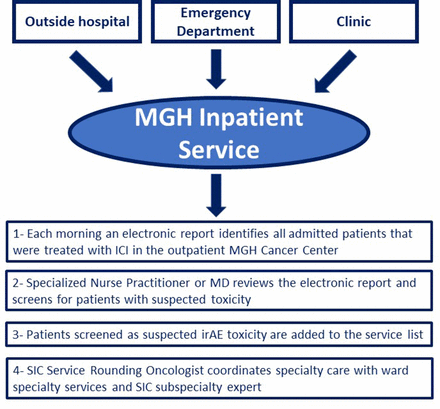
Effect of a multidisciplinary Severe Immunotherapy Complications Service on outcomes for patients receiving immune checkpoint inhibitor therapy for cancer
J Immunother Cancer, 9(9). 2021 Sep.
Abstract
BACKGROUND: In 2017, Massachusetts General Hospital implemented the Severe Immunotherapy Complications (SIC) Service, a multidisciplinary care team for patients hospitalized with immune-related adverse events (irAEs), a unique spectrum of toxicities associated with immune checkpoint inhibitors (ICIs). This study's objectives were to evaluate the intervention's (1) effect on patient outcomes and healthcare utilization, and (2) ability to collect biological samples via a central infrastructure, in order to study the mechanisms responsible for irAEs. METHODS: A hospital database was used to identify patients who received ICIs for a malignancy and were hospitalized with severe irAEs, before (April 2, 2016-October 3, 2017) and after (October 3, 2017-October 24, 2018) SIC Service initiation. The primary outcome was readmission rate after index hospitalization. Secondary outcomes included length of stay (LOS) for admissions, corticosteroid and non-steroidal second-line immunosuppression use, ICI discontinuation, and inpatient mortality. RESULTS: In the pre-SIC period, 127 of 1169 patients treated with ICIs were hospitalized for irAEs; in the post-SIC period, 122 of 1159. After SIC service initiation, reductions were observed in irAE readmission rate (14.8% post-SIC vs 25.9% pre-SIC; OR 0.46; 95% CI 0.22 to 0.95; p=0.036) and readmission LOS (median 6 days post-SIC vs 7 days pre-SIC; 95% CI -16.03 to -0.14; p=0.046). No significant pre-initiation and post-initiation differences were detected in corticosteroid use, second-line immunosuppression, ICI discontinuation, or inpatient mortality rates. The SIC Service collected 789 blood and tissue samples from 234 patients with suspected irAEs. CONCLUSIONS: This is the first study to report that establishing a highly subspecialized care team focused on irAEs is associated with improved patient outcomes and reduced healthcare utilization. Furthermore, the SIC Service successfully integrated blood and tissue collection safety into routine care.
-
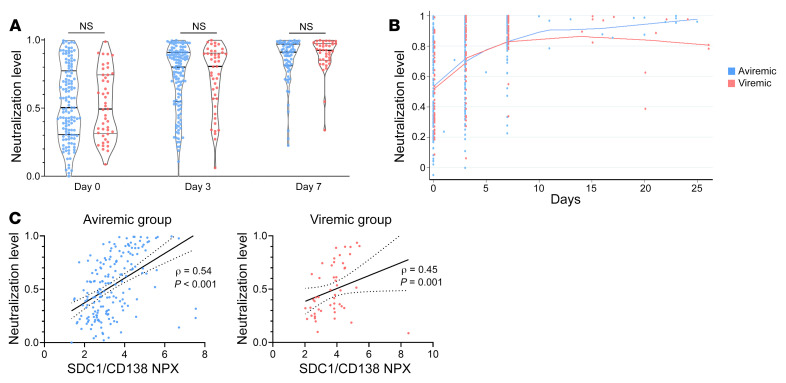
SARS-CoV-2 viremia is associated with distinct proteomic pathways and predicts COVID-19 outcomes
J Clin Invest, 131(13). 2021 Jul 1.
Abstract
BACKGROUNDSARS-CoV-2 plasma viremia has been associated with severe disease and death in COVID-19 in small-scale cohort studies. The mechanisms behind this association remain elusive.METHODSWe evaluated the relationship between SARS-CoV-2 viremia, disease outcome, and inflammatory and proteomic profiles in a cohort of COVID-19 emergency department participants. SARS-CoV-2 viral load was measured using a quantitative reverse transcription PCR-based platform. Proteomic data were generated with Proximity Extension Assay using the Olink platform.RESULTSThis study included 300 participants with nucleic acid test-confirmed COVID-19. Plasma SARS-CoV-2 viremia levels at the time of presentation predicted adverse disease outcomes, with an adjusted OR of 10.6 (95% CI 4.4-25.5, P < 0.001) for severe disease (mechanical ventilation and/or 28-day mortality) and 3.9 (95% CI 1.5-10.1, P = 0.006) for 28-day mortality. Proteomic analyses revealed prominent proteomic pathways associated with SARS-CoV-2 viremia, including upregulation of SARS-CoV-2 entry factors (ACE2, CTSL, FURIN), heightened markers of tissue damage to the lungs, gastrointestinal tract, and endothelium/vasculature, and alterations in coagulation pathways.CONCLUSIONThese results highlight the cascade of vascular and tissue damage associated with SARS-CoV-2 plasma viremia that underlies its ability to predict COVID-19 disease outcomes.FUNDINGMark and Lisa Schwartz; the National Institutes of Health (U19AI082630); the American Lung Association; the Executive Committee on Research at Massachusetts General Hospital; the Chan Zuckerberg Initiative; Arthur, Sandra, and Sarah Irving for the David P. Ryan, MD, Endowed Chair in Cancer Research; an EMBO Long-Term Fellowship (ALTF 486-2018); a Cancer Research Institute/Bristol Myers Squibb Fellowship (CRI2993); the Harvard Catalyst/Harvard Clinical and Translational Science Center (National Center for Advancing Translational Sciences, NIH awards UL1TR001102 and UL1TR002541-01); and by the Harvard University Center for AIDS Research (National Institute of Allergy and Infectious Diseases, 5P30AI060354).
-
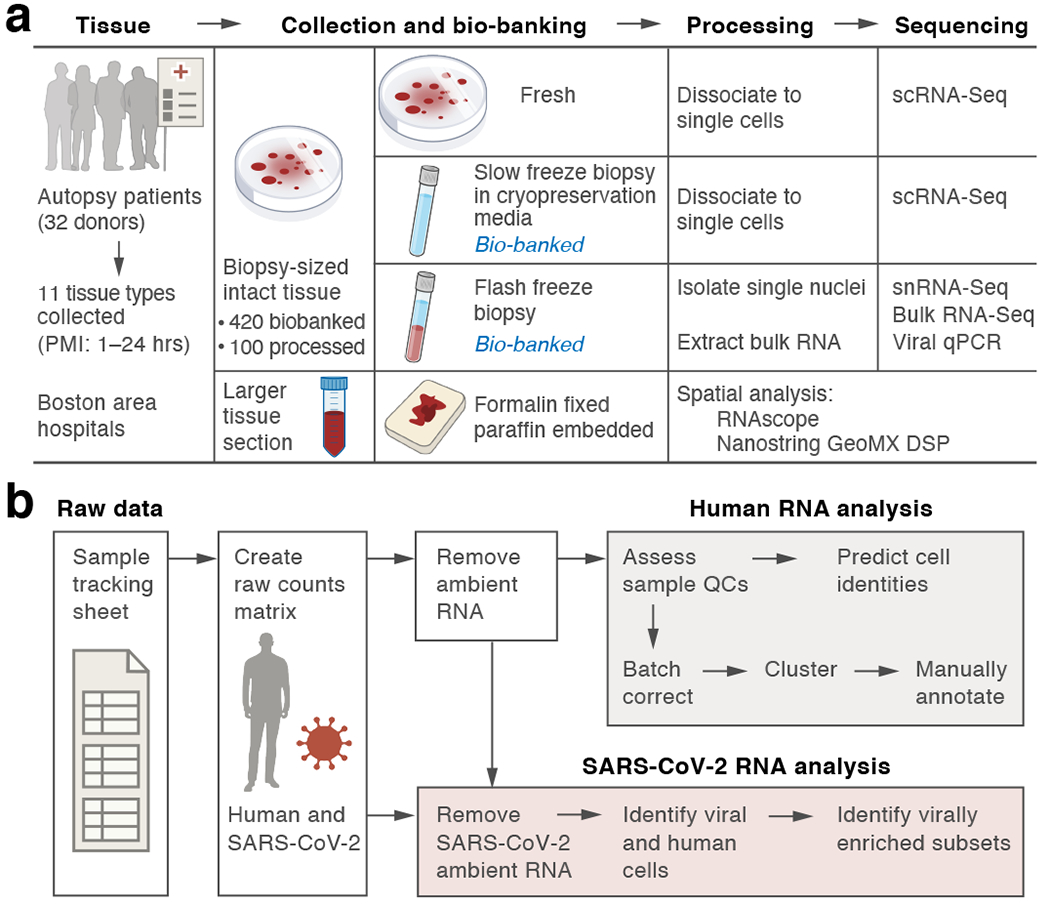
COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets
Nature, 595(7865):107-113. 2021 Jul.
Abstract
COVID-19, which is caused by SARS-CoV-2, can result in acute respiratory distress syndrome and multiple organ failure(1-4), but little is known about its pathophysiology. Here we generated single-cell atlases of 24 lung, 16 kidney, 16 liver and 19 heart autopsy tissue samples and spatial atlases of 14 lung samples from donors who died of COVID-19. Integrated computational analysis uncovered substantial remodelling in the lung epithelial, immune and stromal compartments, with evidence of multiple paths of failed tissue regeneration, including defective alveolar type 2 differentiation and expansion of fibroblasts and putative TP63(+) intrapulmonary basal-like progenitor cells. Viral RNAs were enriched in mononuclear phagocytic and endothelial lung cells, which induced specific host programs. Spatial analysis in lung distinguished inflammatory host responses in lung regions with and without viral RNA. Analysis of the other tissue atlases showed transcriptional alterations in multiple cell types in heart tissue from donors with COVID-19, and mapped cell types and genes implicated with disease severity based on COVID-19 genome-wide association studies. Our foundational dataset elucidates the biological effect of severe SARS-CoV-2 infection across the body, a key step towards new treatments.
-
Immune-related adverse events associated with immune checkpoint inhibitors: a call to action for collecting and sharing clinical trial and real-world data
J Immunother Cancer, 9(7). 2021 Jul.
Abstract
Immune checkpoint inhibitors (ICIs) have revolutionized the treatment of cancer, improving outcomes in patients with advanced malignancies. The use of ICIs in clinical practice, and the number of ICI clinical trials, are rapidly increasing. The use of ICIs in combination with other forms of cancer therapy, such as chemotherapy, radiotherapy, or targeted therapy, is also expanding. However, immune-related adverse events (irAEs) can be serious in up to a third of patients. Critical questions remain surrounding the characteristics and outcomes of irAEs, and how they may affect the overall risk-benefit relationship for combination therapies. This article proposes a framework for irAE classification and reporting, and identifies limitations in the capture and sharing of data on irAEs from current clinical trial and real-world data. We outline key gaps and suggestions for clinicians, clinical investigators, drug sponsors, patients, and other stakeholders to make these critical data more available to researchers for pooled analysis, to advance contemporary understanding of irAEs, and ultimately improve the efficacy of ICIs.
-
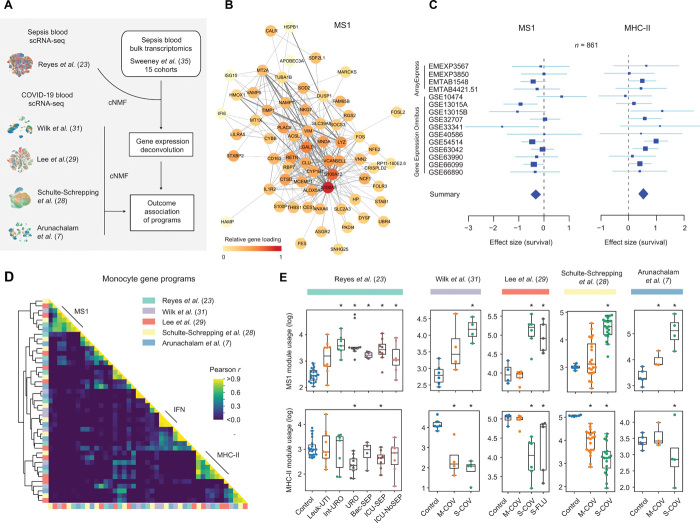
Plasma from patients with bacterial sepsis or severe COVID-19 induces suppressive myeloid cell production from hematopoietic progenitors in vitro
Sci Transl Med, 13(598). 2021 Jun 16.
Abstract
Bacterial sepsis and severe COVID-19 share similar clinical manifestations and are both associated with dysregulation of the myeloid cell compartment. We previously reported an expanded CD14(+) monocyte state, MS1, in patients with bacterial sepsis and validated expansion of this cell subpopulation in publicly available transcriptomics data. Here, using published datasets, we show that the gene expression program associated with MS1 correlated with sepsis severity and was up-regulated in monocytes from patients with severe COVID-19. To examine the ontogeny and function of MS1 cells, we developed a cellular model for inducing CD14(+) MS1 monocytes from healthy bone marrow hematopoietic stem and progenitor cells (HSPCs). We found that plasma from patients with bacterial sepsis or COVID-19 induced myelopoiesis in HSPCs in vitro and expression of the MS1 gene program in monocytes and neutrophils that differentiated from these HSPCs. Furthermore, we found that plasma concentrations of IL-6, and to a lesser extent IL-10, correlated with increased myeloid cell output from HSPCs in vitro and enhanced expression of the MS1 gene program. We validated the requirement for these two cytokines to induce the MS1 gene program through CRISPR-Cas9 editing of their receptors in HSPCs. Using this cellular model system, we demonstrated that induced MS1 cells were broadly immunosuppressive and showed decreased responsiveness to stimulation with a synthetic RNA analog. Our in vitro study suggests a potential role for systemic cytokines in inducing myelopoiesis during severe bacterial or SARS-CoV-2 infection.
-
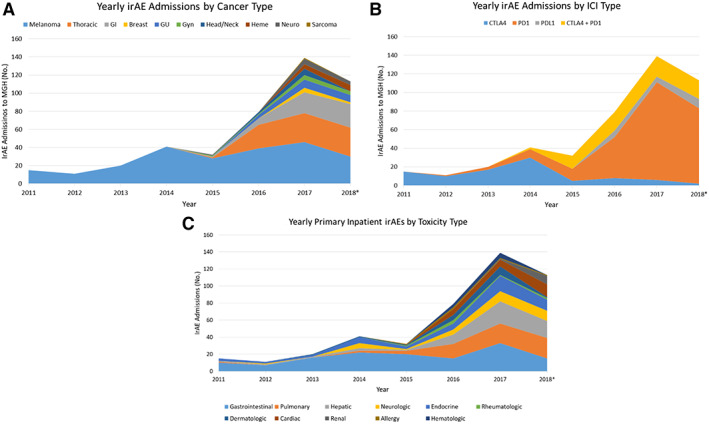
Temporal Trends and Outcomes Among Patients Admitted for Immune-Related Adverse Events: A Single-Center Retrospective Cohort Study from 2011 to 2018
Oncologist, 26(6):514-522. 2021 Jun.
Abstract
BACKGROUND: The aim of this study was to characterize severe immune-related adverse events (irAEs) seen among hospitalized patients and to examine risk factors for irAE admissions and clinically relevant outcomes, including length of stay, immune checkpoint inhibitor (ICI) discontinuation, readmission, and death. METHODS: Patients who received ICI therapy (ipilimumab, pembrolizumab, nivolumab, atezolizumab, durvalumab, avelumab, or any ICI combination) at Massachusetts General Hospital (MGH) and were hospitalized at MGH following ICI initiation between January 1, 2011, and October 24, 2018, were identified using pharmacy and hospital admission databases. Medical records of all irAE admissions were reviewed, and specialist review with defined criteria was performed. Demographic data, relevant clinical history (malignancy type and most recent ICI regimen), and key admission characteristics, including dates of admission and discharge, immunosuppressive management, ICI discontinuation, readmission, and death, were collected. RESULTS: In total, 450 admissions were classified as irAE admissions and represent the study's cohort. Alongside the increasing use of ICIs at our institution, the number of patients admitted to MGH for irAEs has gradually increased every year from 9 in 2011 to 92 in 2018. The hospitalization rate per ICI recipient has declined over that same time period (25.0% in 2011 to 8.5% in 2018). The most common toxicities leading to hospitalization in our cohort were gastrointestinal (30.7%; n = 138), pulmonary (15.8%; n = 71), hepatic (14.2%; n = 64), endocrine (12.2%; n = 55), neurologic (8.4%; n = 38), cardiac (6.7%; n = 30), and dermatologic (4.4%; n = 20). Multivariable logistic regression revealed statistically significant increases in irAE admission risk for CTLA-4 monotherapy recipients (odds ratio [OR], 2.02; p < .001) and CTLA-4 plus PD-1 combination therapy recipients (OR, 1.88; p < .001), relative to PD-1/PD-L1 monotherapy recipients, and patients with multiple toxicity had a 5-fold increase in inpatient mortality. CONCLUSION: This study illustrates that cancer centers must be prepared to manage a wide variety of irAE types and that CTLA-4 and combination ICI regimens are more likely to cause irAE admissions, and earlier. In addition, admissions for patients with multi-organ involvement is common and those patients are at highest risk of inpatient mortality. IMPLICATIONS FOR PRACTICE: The number of patients admitted to Massachusetts General Hospital for immune-related adverse events (irAEs) has gradually increased every year and the most common admissions are for gastrointestinal (30.7%), pulmonary (15/8%), and hepatic (14.2%) events. Readmission rates are high (29% at 30 days, 49% at 180 days) and 64.2% have to permanently discontinue immune checkpoint inhibitor therapy. Importantly, multiple concurrent toxicities were seen in 21.6% (97/450) of irAE admissions and these patients have a fivefold increased risk of inpatient death.
-
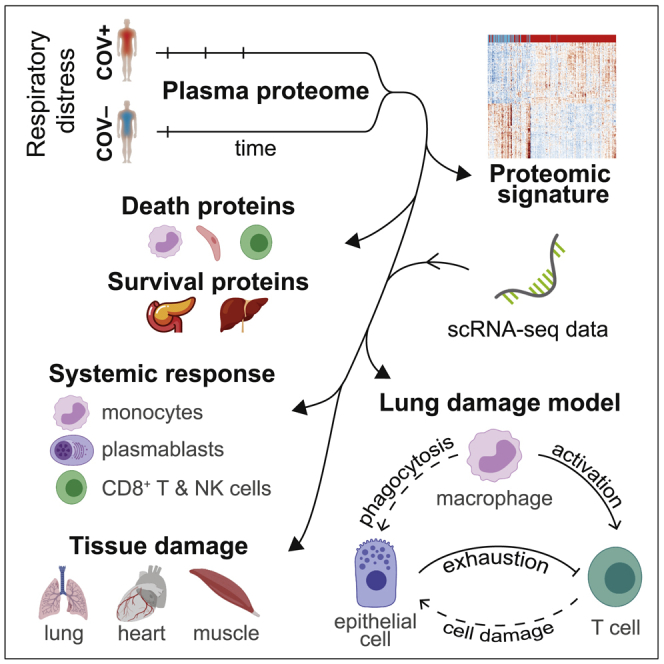
Longitudinal proteomic analysis of severe COVID-19 reveals survival-associated signatures, tissue-specific cell death, and cell-cell interactions
Cell Rep Med, 2(5):100287. 2021 May 18.
Abstract
Mechanisms underlying severe coronavirus disease 2019 (COVID-19) disease remain poorly understood. We analyze several thousand plasma proteins longitudinally in 306 COVID-19 patients and 78 symptomatic controls, uncovering immune and non-immune proteins linked to COVID-19. Deconvolution of our plasma proteome data using published scRNA-seq datasets reveals contributions from circulating immune and tissue cells. Sixteen percent of patients display reduced inflammation yet comparably poor outcomes. Comparison of patients who died to severely ill survivors identifies dynamic immune-cell-derived and tissue-associated proteins associated with survival, including exocrine pancreatic proteases. Using derived tissue-specific and cell-type-specific intracellular death signatures, cellular angiotensin-converting enzyme 2 (ACE2) expression, and our data, we infer whether organ damage resulted from direct or indirect effects of infection. We propose a model in which interactions among myeloid, epithelial, and T cells drive tissue damage. These datasets provide important insights and a rich resource for analysis of mechanisms of severe COVID-19 disease.
-
The Known Unknowns of the Immune Response to Coccidioides
J Fungi (Basel), 7(5). 2021 May 11.
Abstract
Coccidioidomycosis, otherwise known as Valley Fever, is caused by the dimorphic fungi Coccidioides immitis and C. posadasii. While most clinical cases present with self-limiting pulmonary infection, dissemination of Coccidioides spp. results in prolonged treatment and portends higher mortality rates. While the structure, genome, and niches for Coccidioides have provided some insight into the pathogenesis of disease, the underlying immunological mechanisms of clearance or inability to contain the infection in the lung are poorly understood. This review focuses on the known innate and adaptive immune responses to Coccidioides and highlights three important areas of uncertainty and potential approaches to address them. Closing these gaps in knowledge may enable new preventative and therapeutic strategies to be pursued.
-
Widespread haploid-biased gene expression enables sperm-level natural selection
Science, 371(6533). 2021 Mar 5.
Abstract
Sperm are haploid but must be functionally equivalent to distribute alleles equally among progeny. Accordingly, gene products are shared through spermatid cytoplasmic bridges that erase phenotypic differences between individual haploid sperm. Here, we show that a large class of mammalian genes are not completely shared across these bridges. We call these genes "genoinformative markers" (GIMs) and show that a subset can act as selfish genetic elements that spread alleles unevenly through murine, bovine, and human populations. We identify evolutionary pressure to avoid conflict between sperm and somatic function as GIMs are enriched for testis-specific gene expression, paralogs, and isoforms. Therefore, GIMs and sperm-level natural selection may help to explain why testis gene expression patterns are an outlier relative to all other tissues.
-
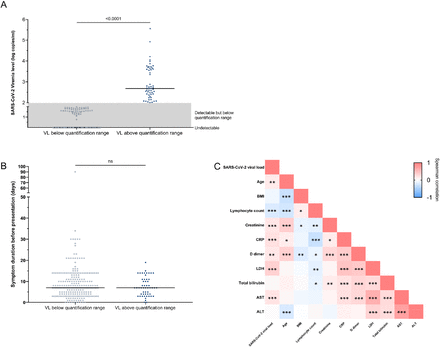
SARS-CoV-2 Viremia is Associated with Distinct Proteomic Pathways and Predicts COVID-19 Outcomes
medRxiv. 2021 Feb 26.
Abstract
BACKGROUND: Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) plasma viremia has been associated with severe disease and death in coronavirus disease 2019 (COVID-19) in small-scale cohort studies. The mechanisms behind this association remain elusive. METHODS: We evaluated the relationship between SARS-CoV-2 viremia, disease outcome, inflammatory and proteomic profiles in a cohort of COVID-19 emergency department participants. SARS-CoV-2 viral load was measured using qRT-PCR based platform. Proteomic data were generated with Proximity Extension Assay (PEA) using the Olink platform. RESULTS: Three hundred participants with nucleic acid test-confirmed COVID-19 were included in this study. Levels of plasma SARS-CoV-2 viremia at the time of presentation predicted adverse disease outcomes, with an adjusted odds ratio (aOR) of 10.6 (95% confidence interval [CI] 4.4, 25.5, P<0.001) for severe disease (mechanical ventilation and/or 28-day mortality) and aOR of 3.9 (95%CI 1.5, 10.1, P=0.006) for 28-day mortality. Proteomic analyses revealed prominent proteomic pathways associated with SARS-CoV-2 viremia, including upregulation of SARS-CoV-2 entry factors (ACE2, CTSL, FURIN), heightened markers of tissue damage to the lungs, gastrointestinal tract, endothelium/vasculature and alterations in coagulation pathways. CONCLUSIONS: These results highlight the cascade of vascular and tissue damage associated with SARS-CoV-2 plasma viremia that underlies its ability to predict COVID-19 disease outcomes.
-

A single-cell and spatial atlas of autopsy tissues reveals pathology and cellular targets of SARS-CoV-2
bioRxiv. 2021 Feb 25.
Abstract
The SARS-CoV-2 pandemic has caused over 1 million deaths globally, mostly due to acute lung injury and acute respiratory distress syndrome, or direct complications resulting in multiple-organ failures. Little is known about the host tissue immune and cellular responses associated with COVID-19 infection, symptoms, and lethality. To address this, we collected tissues from 11 organs during the clinical autopsy of 17 individuals who succumbed to COVID-19, resulting in a tissue bank of approximately 420 specimens. We generated comprehensive cellular maps capturing COVID-19 biology related to patients' demise through single-cell and single-nucleus RNA-Seq of lung, kidney, liver and heart tissues, and further contextualized our findings through spatial RNA profiling of distinct lung regions. We developed a computational framework that incorporates removal of ambient RNA and automated cell type annotation to facilitate comparison with other healthy and diseased tissue atlases. In the lung, we uncovered significantly altered transcriptional programs within the epithelial, immune, and stromal compartments and cell intrinsic changes in multiple cell types relative to lung tissue from healthy controls. We observed evidence of: alveolar type 2 (AT2) differentiation replacing depleted alveolar type 1 (AT1) lung epithelial cells, as previously seen in fibrosis; a concomitant increase in myofibroblasts reflective of defective tissue repair; and, putative TP63(+) intrapulmonary basal-like progenitor (IPBLP) cells, similar to cells identified in H1N1 influenza, that may serve as an emergency cellular reserve for severely damaged alveoli. Together, these findings suggest the activation and failure of multiple avenues for regeneration of the epithelium in these terminal lungs. SARS-CoV-2 RNA reads were enriched in lung mononuclear phagocytic cells and endothelial cells, and these cells expressed distinct host response transcriptional programs. We corroborated the compositional and transcriptional changes in lung tissue through spatial analysis of RNA profiles in situ and distinguished unique tissue host responses between regions with and without viral RNA, and in COVID-19 donor tissues relative to healthy lung. Finally, we analyzed genetic regions implicated in COVID-19 GWAS with transcriptomic data to implicate specific cell types and genes associated with disease severity. Overall, our COVID-19 cell atlas is a foundational dataset to better understand the biological impact of SARS-CoV-2 infection across the human body and empowers the identification of new therapeutic interventions and prevention strategies.
-
Developmental cell programs are co-opted in inflammatory skin disease
Science, 371(6527). 2021 Jan 22.
Abstract
The skin confers biophysical and immunological protection through a complex cellular network established early in embryonic development. We profiled the transcriptomes of more than 500,000 single cells from developing human fetal skin, healthy adult skin, and adult skin with atopic dermatitis and psoriasis. We leveraged these datasets to compare cell states across development, homeostasis, and disease. Our analysis revealed an enrichment of innate immune cells in skin during the first trimester and clonal expansion of disease-associated lymphocytes in atopic dermatitis and psoriasis. We uncovered and validated in situ a reemergence of prenatal vascular endothelial cell and macrophage cellular programs in atopic dermatitis and psoriasis lesional skin. These data illustrate the dynamism of cutaneous immunity and provide opportunities for targeting pathological developmental programs in inflammatory skin diseases.
-
Altered ratio of dendritic cell subsets in skin-draining lymph nodes promotes Th2-driven contact hypersensitivity
Proc Natl Acad Sci U S A, 118(3). 2021 Jan 19.
Abstract
Plasmacytoid dendritic cells (pDCs) specialize in the production of type I IFN (IFN-I). pDCs can be depleted in vivo by injecting diphtheria toxin (DT) in a mouse in which pDCs express a diphtheria toxin receptor (DTR) transgene driven by the human CLEC4C promoter. This promoter is enriched for binding sites for TCF4, a transcription factor that promotes pDC differentiation and expression of pDC markers, including CLEC4C. Here, we found that injection of DT in CLEC4C-DTR(+) mice markedly augmented Th2-dependent skin inflammation in a model of contact hypersensitivity (CHS) induced by the hapten fluorescein isothiocyanate. Unexpectedly, this biased Th2 response was independent of reduced IFN-I accompanying pDC depletion. In fact, DT treatment altered the representation of conventional dendritic cells (cDCs) in the skin-draining lymph nodes during the sensitization phase of CHS; there were fewer Th1-priming CD326(+) CD103(+) cDC1 and more Th2-priming CD11b(+) cDC2. Single-cell RNA-sequencing of CLEC4C-DTR(+) cDCs revealed that CD326(+) DCs, like pDCs, expressed DTR and were depleted together with pDCs by DT treatment. Since CD326(+) DCs did not express Tcf4, DTR expression might be driven by yet-undefined transcription factors activating the CLEC4C promoter. These results demonstrate that altered DC representation in the skin-draining lymph nodes during sensitization to allergens can cause Th2-driven CHS.
-
Antigen Presenting Cells Link the Female Genital Tract Microbiome to Mucosal Inflammation, With Hormonal Contraception as an Additional Modulator of Inflammatory Signatures
Front Cell Infect Microbiol, 11:733619. 2021.
Abstract
The microbiome of the female genital tract (FGT) is closely linked to reproductive health outcomes. Diverse, anaerobe-dominated communities with low Lactobacillus abundance are associated with a number of adverse reproductive outcomes, such as preterm birth, cervical dysplasia, and sexually transmitted infections (STIs), including HIV. Vaginal dysbiosis is associated with local mucosal inflammation, which likely serves as a biological mediator of poor reproductive outcomes. Yet the precise mechanisms of this FGT inflammation remain unclear. Studies in humans have been complicated by confounding demographic, behavioral, and clinical variables. Specifically, hormonal contraception is associated both with changes in the vaginal microbiome and with mucosal inflammation. In this study, we examined the transcriptional landscape of cervical cell populations in a cohort of South African women with differing vaginal microbial community types. We also investigate effects of reproductive hormones on the transcriptional profiles of cervical cells, focusing on the contraceptive depot medroxyprogesterone acetate (DMPA), the most common form of contraception in sub-Saharan Africa. We found that antigen presenting cells (APCs) are key mediators of microbiome associated FGT inflammation. We also found that DMPA is associated with significant transcriptional changes across multiple cell lineages, with some shared and some distinct pathways compared to the inflammatory signature seen with dysbiosis. These results highlight the importance of an integrated, systems-level approach to understanding host-microbe interactions, with an appreciation for important variables, such as reproductive hormones, in the complex system of the FGT mucosa.
2020 (13)
-
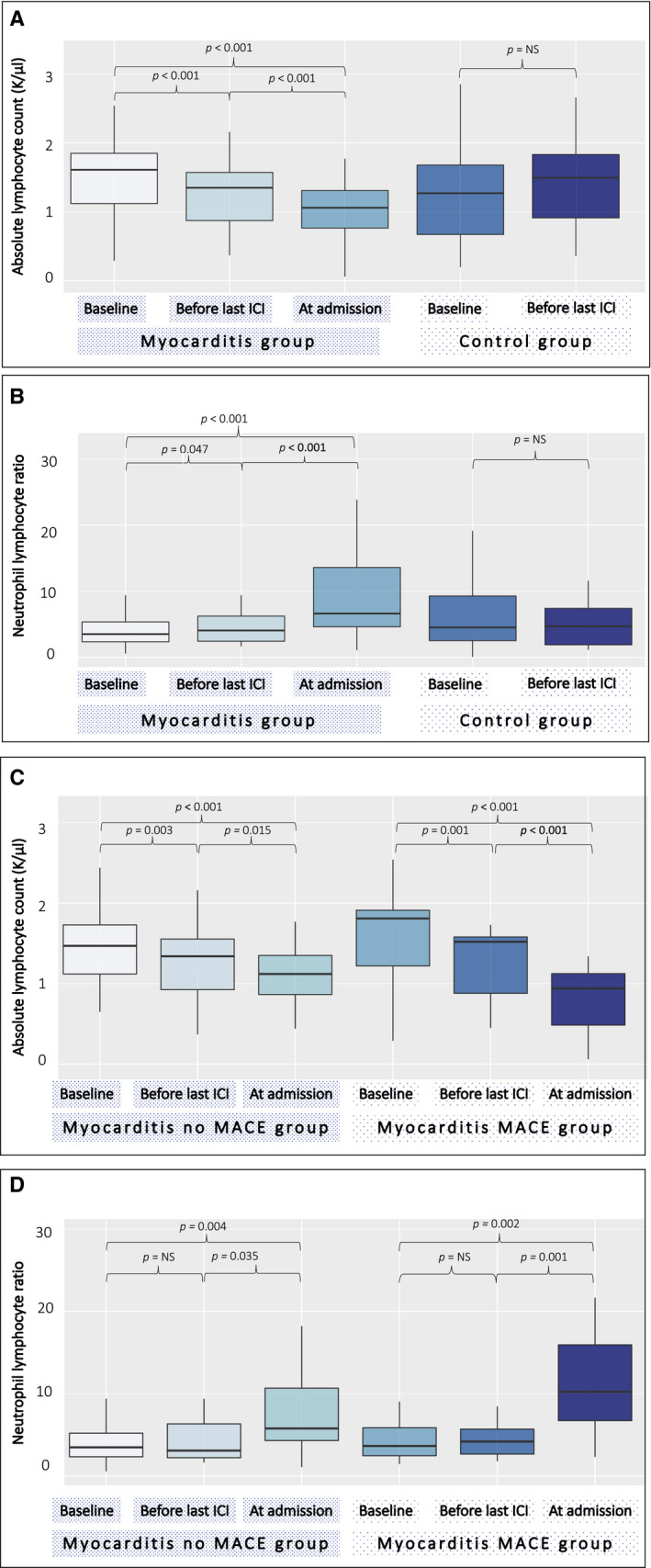
Decreased Absolute Lymphocyte Count and Increased Neutrophil/Lymphocyte Ratio With Immune Checkpoint Inhibitor-Associated Myocarditis
J Am Heart Assoc, 9(23):e018306. 2020 Dec.
Abstract
Background Myocarditis attributable to immune checkpoint inhibitor (ICI) therapy is a potentially fatal immune-related adverse event. Limited data have suggested an association between baseline and on-treatment absolute lymphocyte count (ALC) and neutrophil/lymphocyte ratio (NLR) and the development of other immune-related adverse events; there are no data characterizing the role of ALC and NLR in ICI-associated myocarditis. Methods and Results This was a case control study of 55 patients with ICI myocarditis and 55 controls without any post-ICI immune-related adverse events. We leveraged clinical testing, where patients underwent routine serial blood counts before and with each ICI cycle to compare the baseline and change in ALC and NLR between cases and controls. The association between the change in these parameters with clinical variables and major adverse cardiac events was also tested. In cases, there was a statistically significant decrease in ALC with myocarditis from baseline (1.6 thousands per cubic milliliter (K/μL); interquartile range, 1.1-1.9 K/μL) to admission (1.1 K/μL; interquartile range, 0.7-1.3 K/μL; P<0.001). Similarly, there was an increase in NLR from baseline (3.5; interquartile range, 2.3-5.4) to admission (6.6; interquartile range, 4.5-14.1; P<0.001). There was no statistically significant change in controls. In follow-up, there were 20 events; larger decreases in ALC (44.6% versus 18.2%; P<0.001) or increases in NLR (156.5% versus 65.1%; P=0.019) were associated with major adverse cardiac events. Conclusions A reduction in ALC and an increase in NLR was seen with ICI myocarditis. A greater decrease in ALC or increase in NLR was associated with subsequent major adverse cardiac events.
-
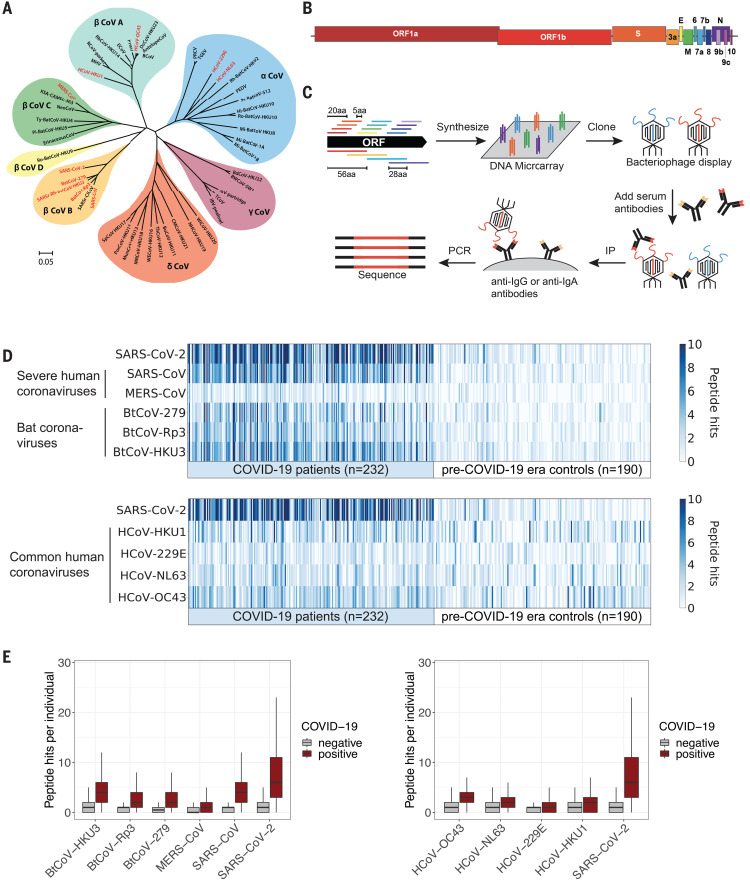
Viral epitope profiling of COVID-19 patients reveals cross-reactivity and correlates of severity
Science, 370(6520). 2020 Nov 27.
Abstract
Understanding humoral responses to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is critical for improving diagnostics, therapeutics, and vaccines. Deep serological profiling of 232 coronavirus disease 2019 (COVID-19) patients and 190 pre-COVID-19 era controls using VirScan revealed more than 800 epitopes in the SARS-CoV-2 proteome, including 10 epitopes likely recognized by neutralizing antibodies. Preexisting antibodies in controls recognized SARS-CoV-2 ORF1, whereas only COVID-19 patient antibodies primarily recognized spike protein and nucleoprotein. A machine learning model trained on VirScan data predicted SARS-CoV-2 exposure history with 99% sensitivity and 98% specificity; a rapid Luminex-based diagnostic was developed from the most discriminatory SARS-CoV-2 peptides. Individuals with more severe COVID-19 exhibited stronger and broader SARS-CoV-2 responses, weaker antibody responses to prior infections, and higher incidence of cytomegalovirus and herpes simplex virus 1, possibly influenced by demographic covariates. Among hospitalized patients, males produce stronger SARS-CoV-2 antibody responses than females.
-
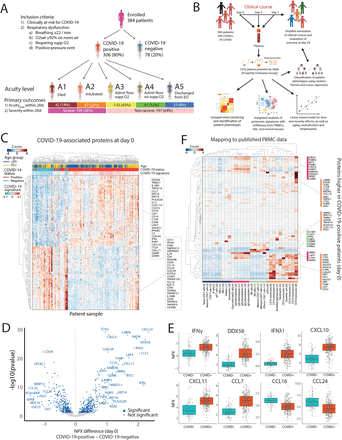
Plasma proteomics reveals tissue-specific cell death and mediators of cell-cell interactions in severe COVID-19 patients
bioRxiv. 2020 Nov 4.
Abstract
COVID-19 has caused over 1 million deaths globally, yet the cellular mechanisms underlying severe disease remain poorly understood. By analyzing several thousand plasma proteins in 306 COVID-19 patients and 78 symptomatic controls over serial timepoints using two complementary approaches, we uncover COVID-19 host immune and non-immune proteins not previously linked to this disease. Integration of plasma proteomics with nine published scRNAseq datasets shows that SARS-CoV-2 infection upregulates monocyte/macrophage, plasmablast, and T cell effector proteins. By comparing patients who died to severely ill patients who survived, we identify dynamic immunomodulatory and tissue-associated proteins associated with survival, providing insights into which host responses are beneficial and which are detrimental to survival. We identify intracellular death signatures from specific tissues and cell types, and by associating these with angiotensin converting enzyme 2 (ACE2) expression, we map tissue damage associated with severe disease and propose which damage results from direct viral infection rather than from indirect effects of illness. We find that disease severity in lung tissue is driven by myeloid cell phenotypes and cell-cell interactions with lung epithelial cells and T cells. Based on these results, we propose a model of immune and epithelial cell interactions that drive cell-type specific and tissue-specific damage in severe COVID-19.
-
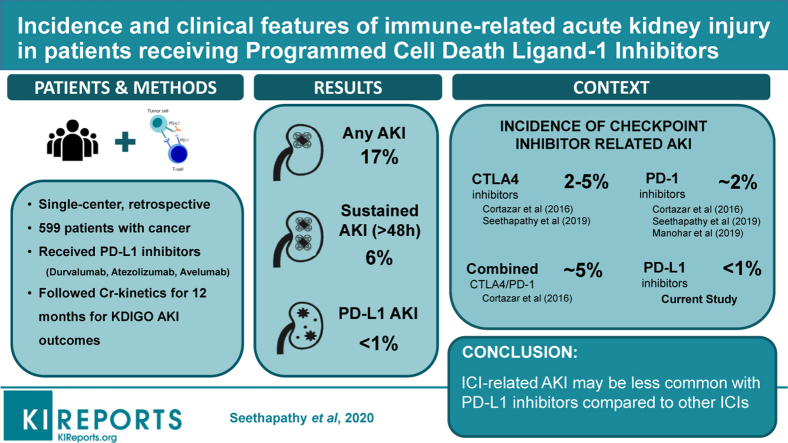
Incidence and Clinical Features of Immune-Related Acute Kidney Injury in Patients Receiving Programmed Cell Death Ligand-1 Inhibitors
Kidney Int Rep, 5(10):1700-1705. 2020 Oct.
Abstract
BACKGROUND: Programmed cell death receptor ligand 1 (PD-L1) inhibitors are immune checkpoint inhibitors (ICIs) with a side effect profile that may differ from other classes of ICIs such as those directed against cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death 1 receptor (PD-1). Being the more recently approved class of checkpoint inhibitors, there are no studies investigating the frequency, etiology and predictors of acute kidney injury (AKI) in patients receiving PD-L1 inhibitors. METHODS: This was a retrospective cohort study of patients who received PD-L1 inhibitors during 2017 to 2018 in our healthcare system. AKI was defined by a ≥1.5-fold rise in serum creatinine from baseline. The etiology of all cases of sustained AKI (lasting >48 hours) and clinical course were determined by review of electronic health records. RESULTS: The final analysis included 599 patients. Within 12 months of ICI initiation, 104 patients (17%) experienced AKI, and 36 (6%) experienced sustained AKI; however, only 5 (<1%) experienced suspected PD-L1-related AKI. The PD-L1-related AKI occurred a median of 99 days after starting therapy. All patients concurrently received another medication known to cause acute interstitial nephritis (proton pump inhibitors, nonsteroidal anti-inflammatory drugs, or antibiotics) at the time of the suspected PDL1-related AKI. CONCLUSION: Although AKI is common in patients receiving PD-L1 therapy, the incidence of suspected PD-L1-related AKI is low (<1%) and may be less common when compared to other classes of ICIs. This cohort provides further validation that other drugs associated with acute interstitial nephritis may be involved in the pathogenesis of ICI-related AKI.
-
Induction of a regulatory myeloid program in bacterial sepsis and severe COVID-19
bioRxiv. 2020 Sep 2.
Abstract
A recent estimate suggests that one in five deaths globally are associated with sepsis (1) . To date, no targeted treatment is available for this syndrome, likely due to substantial patient heterogeneity (2,3) and our lack of insight into sepsis immunopathology (4) . These issues are highlighted by the current COVID-19 pandemic, wherein many clinical manifestations of severe SARS-CoV-2 infection parallel bacterial sepsis (5-8) . We previously reported an expanded CD14+ monocyte state, MS1, in patients with bacterial sepsis or non-infectious critical illness, and validated its expansion in sepsis across thousands of patients using public transcriptomic data (9) . Despite its marked expansion in the circulation of bacterial sepsis patients, its relevance to viral sepsis and association with disease outcomes have not been examined. In addition, the ontogeny and function of this monocyte state remain poorly characterized. Using public transcriptomic data, we show that the expression of the MS1 program is associated with sepsis mortality and is up-regulated in monocytes from patients with severe COVID-19. We found that blood plasma from bacterial sepsis or COVID-19 patients with severe disease induces emergency myelopoiesis and expression of the MS1 program, which are dependent on the cytokines IL-6 and IL-10. Finally, we demonstrate that MS1 cells are broadly immunosuppressive, similar to monocytic myeloid-derived suppressor cells (MDSCs), and have decreased responsiveness to stimulation. Our findings highlight the utility of regulatory myeloid cells in sepsis prognosis, and the role of systemic cytokines in inducing emergency myelopoiesis during severe bacterial and SARS-CoV-2 infections.
-

The activation trajectory of plasmacytoid dendritic cells in vivo during a viral infection
Nat Immunol, 21(9):983-997. 2020 Sep.
Abstract
Plasmacytoid dendritic cells (pDCs) are a major source of type I interferon (IFN-I). What other functions pDCs exert in vivo during viral infections is controversial, and more studies are needed to understand their orchestration. In the present study, we characterize in depth and link pDC activation states in animals infected by mouse cytomegalovirus by combining Ifnb1 reporter mice with flow cytometry, single-cell RNA sequencing, confocal microscopy and a cognate CD4 T cell activation assay. We show that IFN-I production and T cell activation were performed by the same pDC, but these occurred sequentially in time and in different micro-anatomical locations. In addition, we show that pDC commitment to IFN-I production was marked early on by their downregulation of leukemia inhibitory factor receptor and was promoted by cell-intrinsic tumor necrosis factor signaling. We propose a new model for how individual pDCs are endowed to exert different functions in vivo during a viral infection, in a manner tightly orchestrated in time and space.
-
Cumulus provides cloud-based data analysis for large-scale single-cell and single-nucleus RNA-seq
Nat Methods, 17(8):793-798. 2020 Aug.
Abstract
Massively parallel single-cell and single-nucleus RNA sequencing has opened the way to systematic tissue atlases in health and disease, but as the scale of data generation is growing, so is the need for computational pipelines for scaled analysis. Here we developed Cumulus-a cloud-based framework for analyzing large-scale single-cell and single-nucleus RNA sequencing datasets. Cumulus combines the power of cloud computing with improvements in algorithm and implementation to achieve high scalability, low cost, user-friendliness and integrated support for a comprehensive set of features. We benchmark Cumulus on the Human Cell Atlas Census of Immune Cells dataset of bone marrow cells and show that it substantially improves efficiency over conventional frameworks, while maintaining or improving the quality of results, enabling large-scale studies.
-
Systematic comparison of single-cell and single-nucleus RNA-sequencing methods
Nat Biotechnol, 38(6):737-746. 2020 Jun.
Abstract
The scale and capabilities of single-cell RNA-sequencing methods have expanded rapidly in recent years, enabling major discoveries and large-scale cell mapping efforts. However, these methods have not been systematically and comprehensively benchmarked. Here, we directly compare seven methods for single-cell and/or single-nucleus profiling-selecting representative methods based on their usage and our expertise and resources to prepare libraries-including two low-throughput and five high-throughput methods. We tested the methods on three types of samples: cell lines, peripheral blood mononuclear cells and brain tissue, generating 36 libraries in six separate experiments in a single center. To directly compare the methods and avoid processing differences introduced by the existing pipelines, we developed scumi, a flexible computational pipeline that can be used with any single-cell RNA-sequencing method. We evaluated the methods for both basic performance, such as the structure and alignment of reads, sensitivity and extent of multiplets, and for their ability to recover known biological information in the samples.
-
-
Transcriptomic Analysis and High-dimensional Phenotypic Mapping of Mononuclear Phagocytes in Mesenteric Lymph Nodes Reveal Differences Between Ulcerative Colitis and Crohn's Disease
J Crohns Colitis, 14(3):393-405. 2020 Mar 13.
Abstract
BACKGROUND AND AIMS: Crohn's disease [CD] and ulcerative colitis [UC] are distinct forms of inflammatory bowel disease. Heterogeneity of HLA-DR+SIRPα + mononuclear phagocytes [MNPs], including macrophages [MΦ], monocyte-derived [Mono] cells, and dendritic cells [DCs], was reported in gut tissue but not yet investigated in mesenteric lymph nodes [MLNs] of IBD patients. We here compared the phenotype, function, and molecular profile of HLA-DR+SIRPα + MNPs in CD and UC MLNs. METHODS: Cell distribution, morphology, immune function, and transcriptomic [bulk RNAseq] and high-dimensional protein expression profiles [CyTOF] of HLA-DR+SIRPα + MNPs were examined in MLNs of UC [n = 14], CD [n = 35], and non-IBD [n = 12] patients. RESULTS: Elevated frequencies of CD14+CD64+CD163+ [Mono/MΦ-like] MNPs displaying monocyte/MΦ morphology and phagocytic function were a distinct feature of UC MLNs. In CD, the proportion of CD14-CD64-CD163- [DC-like] cells was augmented relative to Mono/MΦ-like cells; DC-like cells drove naïve T cell proliferation, Th1 polarisation, and Th17 TCM plasticity. Gene expression profile corroborated the nature of DC-like cells, best represented by BTLA, SERPINF, IGJ and, of Mono/MΦ-like cells, defined by CD163, MARCO, MAFB, CD300E, S100A9 expression. CyTOF analysis showed that CD123+ plasmacytoid cells predominated over conventional DCs in DC-like cells. Four CD163+ clusters were revealed in Mono/MΦ-like cells, two of which were enriched in MARCO-CD68dimHLA-DRdim monocyte-like cells and MARCOhiCD68hiHLA-DRhi Mɸ, whose proportion increased in UC relative to CD. CONCLUSIONS: Defining the landscape of MNPs in MLNs provided evidence for expansion of CD163+ Mono/MΦ-like cells in UC only, highlighting a distinction between UC and CD, and thus the potential contribution of monocyte-like cells in driving colitis.
-
SLAMF6 deficiency augments tumor killing and skews toward an effector phenotype revealing it as a novel T cell checkpoint
Elife, 9. 2020 Mar 3.
Abstract
SLAMF6 is a homotypic receptor of the Ig-superfamily whose exact role in immune modulation has remained elusive. Its constitutive expression on resting and activated T cells precludes it from being a bona fide exhaustion marker. By breeding Pmel-1 mice with SLAMF6 -/- mice, we generated donors for T cells lacking SLAMF6 and expressing a transgenic TCR for gp100-melanoma antigen. Activated Pmel-1xSLAMF6 -/- CD8+ T cells displayed improved polyfunctionality and strong tumor cytolysis. T-bet was the dominant transcription factor in Pmel-1 x SLAMF6 -/- cells, and upon activation, they acquired an effector-memory phenotype. Adoptive transfer of Pmel-1 x SLAMF6 -/- T cells to melanoma-bearing mice resulted in lasting tumor regression in contrast to temporary responses achieved with Pmel-1 T cells. LAG-3 expression was elevated in the SLAMF6 -/- cells, and the addition of the LAG-3-blocking antibody to the adoptive transfer protocol improved the SLAMF6 -/- T cells and expedited the antitumor response even further. The results from this study support the notion that SLAMF6 is an inhibitory immune receptor whose absence enables powerful CD8+ T cells to eradicate tumors.
-
-
IL-12 and Mucosal CD14+ Monocyte-Like Cells Induce IL-8 in Colonic Memory CD4+ T Cells of Patients With Ulcerative Colitis but not Crohn's Disease
J Crohns Colitis, 14(1):79-95. 2020 Jan 1.
Abstract
BACKGROUND AND AIMS: CD14+ mononuclear phagocytes [MNPs] and T cells infiltrate colon in ulcerative colitis [UC]. Here we investigated how CD14+ MNPs and the cytokines they produce shape the colonic effector T cell profile. METHODS: Colonic or mesenteric lymph node [mLNs] CD4+ T cells isolated from UC or Crohn's disease [CD] patients were stimulated with cytokines or autologous CD14+ MNPs. Cytokine expression was assessed by intracytoplasmic staining and multiplex ELISA. Unsupervised phenotypic multicolour analysis of colonic CD14+ MNPs was performed using the FlowSOM algorithm. RESULTS: Among CD14+CD64+HLA-DR+SIRPα + MNPs, only the pro-inflammatory cytokine-producing CD163- subpopulation accumulated in inflamed UC colon and promoted mucosal IL-1β-dependent Th17, Th17/Th1, Th17/Th22 but not Th1 responses. Unsupervised phenotypic analysis of CD14+CD64+ MNPs segregated CD163- monocyte-like cells and CD163+ macrophages. Unexpectedly, IL-12, IL-1β and CD163-, but not CD163+, cells induced IL-8 expression in colonic CD4+ T cells, which co-expressed IFN-γ and/or IL-17 in UC and not CD. The CD163- monocyte-like cells increased the frequency of IL-8+IL-17+/-IFN-γ +/- T cells through IL-1β and IL-12. Finally, colonic IL-8+ T cells co-expressing GM-CSF, TNF-α and IL-6 were detected ex vivo and, promoted by IL-12 in the mucosa and mLNs in UC only. CONCLUSIONS: Our findings established a link between monocyte-like CD163- MNPs, IL-12, IL-1β and the detection of colonic memory IL-8-producing CD4+ T cells, which might all contribute to the pathogenesis of UC.
2019 (8)
-

The Incidence, Causes, and Risk Factors of Acute Kidney Injury in Patients Receiving Immune Checkpoint Inhibitors
Clin J Am Soc Nephrol, 14(12):1692-1700. 2019 Dec 6.
Abstract
BACKGROUND AND OBJECTIVES: Immune checkpoint inhibitor use in oncology is increasing rapidly. We sought to determine the frequency, severity, cause, and predictors of AKI in a real-world population receiving checkpoint inhibitors. DESIGN, SETTING, PARTICIPANTS, & MEASUREMENTS: We included all patients who received checkpoint inhibitor therapy from May 2011 to December 2016 at Massachusetts General Hospital. Baseline serum creatinine, averaged 6 months before checkpoint inhibitor start date, was compared with all subsequent creatinine values within 12 months of starting therapy. AKI was defined by Kidney Disease: Improving Global Outcomes criteria for fold changes in creatinine from baseline. Sustained AKI events lasted at least 3 days and was our primary outcome. The cause of sustained AKI was determined by chart review. Cumulative incidence and subdistribution hazard models were used to assess the relationship between baseline demographics, comorbidities, and medications, and sustained AKI and potential checkpoint inhibitor-related AKI. RESULTS: We included 1016 patients in the analysis. Average age was 63 (SD 13) years, 61% were men, and 91% were white. Mean baseline creatinine was 0.9 mg/dl (SD 0.4 mg/dl), and 169 (17%) had CKD (eGFR<60 ml/min per 1.73 m(2)) at baseline. A total of 169 patients (17%) experienced AKI, defined by an increase in creatinine at least 1.5 times the baseline within 12 months; 82 patients (8%) experienced sustained AKI and 30 patients (3%) had potential checkpoint inhibitor-related AKI. The first episode of sustained AKI occurred, on average, 106 days (SD 85) after checkpoint inhibitor initiation. Sixteen (2%) patients experienced stage 3 sustained AKI and four patients required dialysis. Proton pump inhibitor use at baseline was associated with sustained AKI. CONCLUSIONS: AKI is common in patients receiving checkpoint inhibitor therapy. The causes of sustained AKI in this population are heterogenous and merit thorough evaluation. The role of PPI and other nephritis-inducing drugs in the development of sustained AKI needs to be better defined.
-
Decoding human fetal liver haematopoiesis
Nature, 574(7778):365-371. 2019 Oct.
Abstract
Definitive haematopoiesis in the fetal liver supports self-renewal and differentiation of haematopoietic stem cells and multipotent progenitors (HSC/MPPs) but remains poorly defined in humans. Here, using single-cell transcriptome profiling of approximately 140,000 liver and 74,000 skin, kidney and yolk sac cells, we identify the repertoire of human blood and immune cells during development. We infer differentiation trajectories from HSC/MPPs and evaluate the influence of the tissue microenvironment on blood and immune cell development. We reveal physiological erythropoiesis in fetal skin and the presence of mast cells, natural killer and innate lymphoid cell precursors in the yolk sac. We demonstrate a shift in the haemopoietic composition of fetal liver during gestation away from being predominantly erythroid, accompanied by a parallel change in differentiation potential of HSC/MPPs, which we functionally validate. Our integrated map of fetal liver haematopoiesis provides a blueprint for the study of paediatric blood and immune disorders, and a reference for harnessing the therapeutic potential of HSC/MPPs.
-
Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis
Cell, 178(3):714-730.e22. 2019 Jul 25.
Abstract
Genome-wide association studies (GWAS) have revealed risk alleles for ulcerative colitis (UC). To understand their cell type specificities and pathways of action, we generate an atlas of 366,650 cells from the colon mucosa of 18 UC patients and 12 healthy individuals, revealing 51 epithelial, stromal, and immune cell subsets, including BEST4(+) enterocytes, microfold-like cells, and IL13RA2(+)IL11(+) inflammatory fibroblasts, which we associate with resistance to anti-TNF treatment. Inflammatory fibroblasts, inflammatory monocytes, microfold-like cells, and T cells that co-express CD8 and IL-17 expand with disease, forming intercellular interaction hubs. Many UC risk genes are cell type specific and co-regulated within relatively few gene modules, suggesting convergence onto limited sets of cell types and pathways. Using this observation, we nominate and infer functions for specific risk genes across GWAS loci. Our work provides a framework for interrogating complex human diseases and mapping risk variants to cell types and pathways.
-
Cardiotoxicity of Immune Checkpoint Inhibitors
Curr Treat Options Cardiovasc Med, 21(7):32. 2019 Jun 8.
Abstract
PURPOSE OF REVIEW: Immunotherapies, particularly immune checkpoint inhibitors (ICI), are revolutionary cancer therapies being increasingly applied to a broader range of cancers. Our understanding of the mechanism, epidemiology, diagnosis, and treatment of cardiotoxicity related to immunotherapies remains limited. We aim to synthesize the limited current literature on cardiotoxicity of ICIs and to share our opinions on the diagnosis and treatment of this condition. RECENT FINDINGS: The incidence of ICI-associated myocarditis ranges from 0.1 to 1%. Patients with ICI-associated myocarditis often have a fulminant course with a case fatality rate of 25-50%. The diagnosis of this condition poses many challenges because independently a normal electrocardiogram, biomarkers, or a preserved left ventricular function do not rule out ICI-associated myocarditis. Endomyocardial biopsy should be pursued when clinical suspicion remains despite normal non-invasive tests. Data on optimal screening and surveillance tools are lacking. Cessation of ICIs, combined with high dose corticosteroids and other immunosuppressant approaches are the cornerstones of the treatment of ICI-associated myocarditis. This condition may recur when patients are re-challenged with these agents and the decision to resume ICIs should be made through a multidisciplinary discussion. Immunotherapies have changed the landscape of cancer treatment. Recognizing and managing cardiotoxicity related to ICIs is of critical importance. Our understanding of ICI-cardiotoxicity has improved, but large information gaps remain for further research. Due to the high case fatality rate, any type of cardiac symptoms or signs in a patient who has recently started an ICI should prompt consideration of ICI-cardiotoxicity.
-

Targeting the CBM complex causes T(reg) cells to prime tumours for immune checkpoint therapy
Nature, 570(7759):112-116. 2019 Jun.
Abstract
Solid tumours are infiltrated by effector T cells with the potential to control or reject them, as well as by regulatory T (T(reg)) cells that restrict the function of effector T cells and thereby promote tumour growth(1). The anti-tumour activity of effector T cells can be therapeutically unleashed, and is now being exploited for the treatment of some forms of human cancer. However, weak tumour-associated inflammatory responses and the immune-suppressive function of T(reg) cells remain major hurdles to broader effectiveness of tumour immunotherapy(2). Here we show that, after disruption of the CARMA1-BCL10-MALT1 (CBM) signalosome complex, most tumour-infiltrating T(reg) cells produce IFNγ, resulting in stunted tumour growth. Notably, genetic deletion of both or even just one allele of CARMA1 (also known as Card11) in only a fraction of T(reg) cells-which avoided systemic autoimmunity-was sufficient to produce this anti-tumour effect, showing that it is not the mere loss of suppressive function but the gain of effector activity by T(reg) cells that initiates tumour control. The production of IFNγ by T(reg) cells was accompanied by activation of macrophages and upregulation of class I molecules of the major histocompatibility complex on tumour cells. However, tumour cells also upregulated the expression of PD-L1, which indicates activation of adaptive immune resistance(3). Consequently, blockade of PD-1 together with CARMA1 deletion caused rejection of tumours that otherwise do not respond to anti-PD-1 monotherapy. This effect was reproduced by pharmacological inhibition of the CBM protein MALT1. Our results demonstrate that partial disruption of the CBM complex and induction of IFNγ secretion in the preferentially self-reactive T(reg) cell pool does not cause systemic autoimmunity but is sufficient to prime the tumour environment for successful immune checkpoint therapy.
-
Two distinct colonic CD14(+) subsets characterized by single-cell RNA profiling in Crohn's disease
Mucosal Immunol, 12(3):703-719. 2019 May.
Abstract
Inflammatory bowel diseases are associated with dysregulated immune responses in the intestinal tissue. Four molecularly identified macrophage subsets control immune homeostasis in healthy gut. However, the specific roles and transcriptomic profiles of the phenotypically heterogeneous CD14(+) macrophage-like population in inflamed gut remain to be investigated in Crohn's disease (CD). Here we identified two phenotypically, morphologically and functionally distinct colonic HLADR(+)SIRPα(+)CD14(+) subpopulations that were further characterized using single-cell RNA-sequencing (scRNAseq) in CD. Frequencies of CD64(hi)CD163(-/dim) cells selectively augmented in inflamed colon and correlated with endoscopic score of disease severity. IL-1β and IL-23-producing CD64(hi)CD163(-/dim) cells predominated over TNF-α-producing CD64(hi)CD163(hi) cells in lesions. Purified "inflammatory monocyte-like" CD163(-), but not "macrophage-like" CD163(hi) cells, through IL-1β, promoted Th17/Th1 but not Th1 responses in tissue memory CD4(+)T cells. Unsupervised scRNAseq analysis that captures the entire HLADR(+)SIRPα(+) population revealed six clusters, two of which were enriched in either CD163(-) or CD163(hi) cells, and best defined by TREM1/FCAR/FCN1/IL1RN or CD209/MERTK/MRCI/CD163L1 genes, respectively. Selected newly identified discriminating markers were used beyond CD163 to isolate cells that shared pro-Th17/Th1 function with CD163(-) cells. In conclusion, a molecularly distinct pro-inflammatory CD14(+) subpopulation accumulates in inflamed colon, drives intestinal inflammatory T-cell responses, and thus, might contribute to CD disease severity.
-
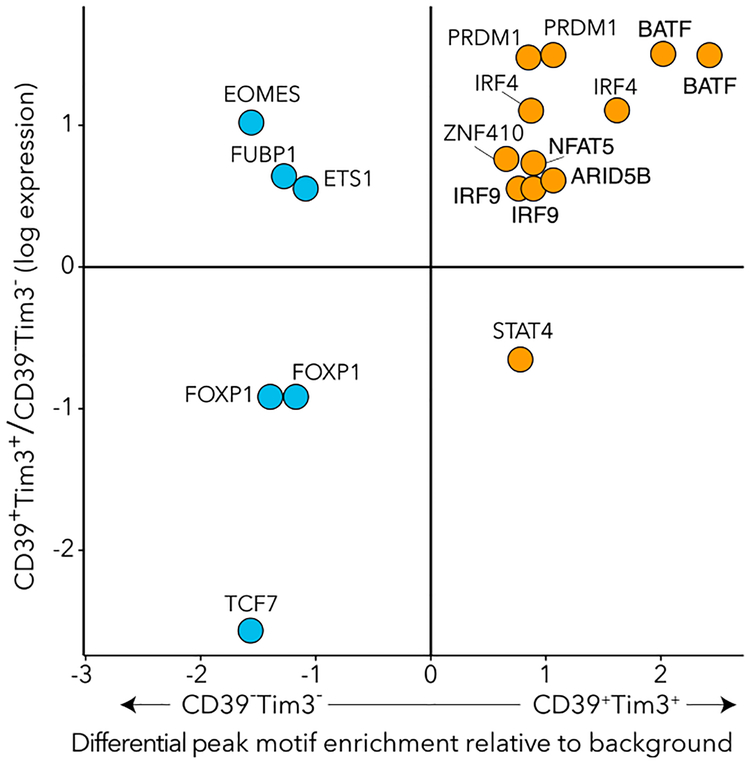
-
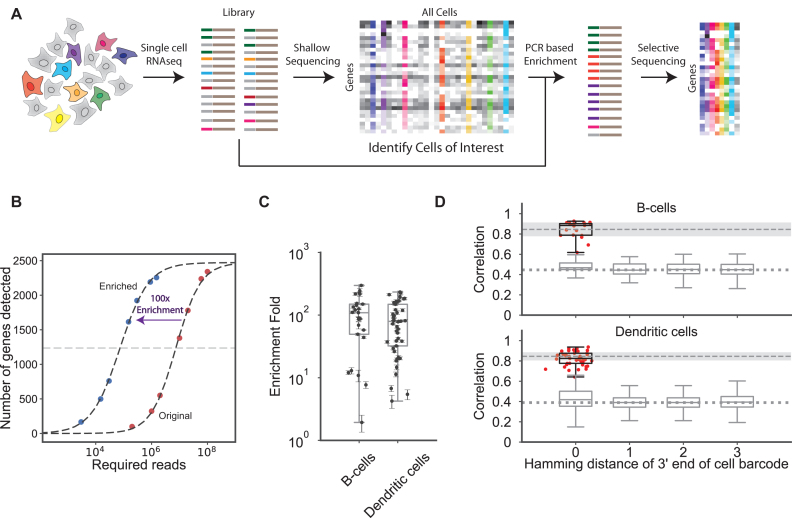
Targeting individual cells by barcode in pooled sequence libraries
Nucleic Acids Res, 47(1):e4. 2019 Jan 10.
Abstract
Transcriptional profiling of thousands of single cells in parallel by RNA-seq is now routine. However, due to reliance on pooled library preparation, targeting analysis to particular cells of interest is difficult. Here, we present a multiplexed PCR method for targeted sequencing of select cells from pooled single-cell sequence libraries. We demonstrated this molecular enrichment method on multiple cell types within pooled single-cell RNA-seq libraries produced from primary human blood cells. We show how molecular enrichment can be combined with FACS to efficiently target ultra-rare cell types, such as the recently identified AXL+SIGLEC6+ dendritic cell (AS DC) subset, in order to reduce the required sequencing effort to profile single cells by 100-fold. Our results demonstrate that DNA barcodes identifying cells within pooled sequencing libraries can be used as targets to enrich for specific molecules of interest, for example reads from a set of target cells.
2018 (6)
-
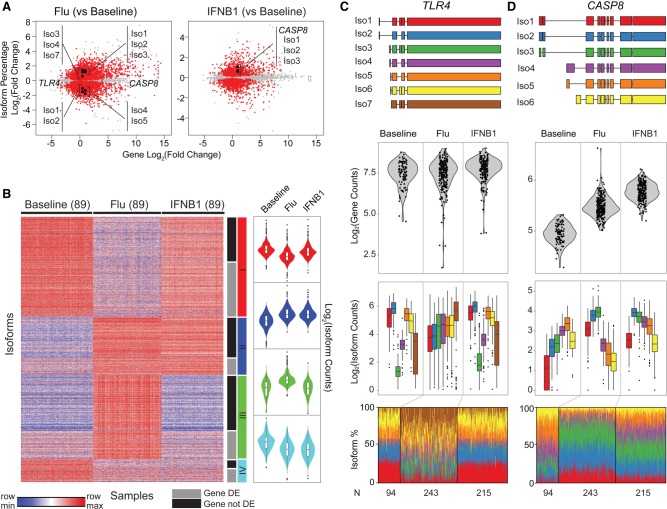
Genetic analysis of isoform usage in the human anti-viral response reveals influenza-specific regulation of ERAP2 transcripts under balancing selection
Genome Res, 28(12):1812-1825. 2018 Dec.
Abstract
While genetic variants are known to be associated with overall gene abundance in stimulated immune cells, less is known about their effects on alternative isoform usage. By analyzing RNA-seq profiles of monocyte-derived dendritic cells from 243 individuals, we uncovered thousands of unannotated isoforms synthesized in response to influenza infection and type 1 interferon stimulation. We identified more than a thousand quantitative trait loci (QTLs) associated with alternate isoform usage (isoQTLs), many of which are independent of expression QTLs (eQTLs) for the same gene. Compared with eQTLs, isoQTLs are enriched for splice sites and untranslated regions, but depleted of sequences upstream of annotated transcription start sites. Both eQTLs and isoQTLs explain a significant proportion of the disease heritability attributed to common genetic variants. At the ERAP2 locus, we shed light on the function of the gene and how two frequent, highly differentiated haplotypes with intermediate frequencies could be maintained by balancing selection. At baseline and following type 1 interferon stimulation, the major haplotype is associated with low ERAP2 expression caused by nonsense-mediated decay, while the minor haplotype, known to increase Crohn's disease risk, is associated with high ERAP2 expression. In response to influenza infection, we found two uncharacterized isoforms expressed from the major haplotype, likely the result of multiple perfectly linked variants affecting the transcription and splicing at the locus. Thus, genetic variants at a single locus could modulate independent gene regulatory processes in innate immune responses and, in the case of ERAP2, may confer a historical fitness advantage in response to virus.
-

Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma
Cell, 175(4):998-1013.e20. 2018 Nov 1.
Abstract
Treatment of cancer has been revolutionized by immune checkpoint blockade therapies. Despite the high rate of response in advanced melanoma, the majority of patients succumb to disease. To identify factors associated with success or failure of checkpoint therapy, we profiled transcriptomes of 16,291 individual immune cells from 48 tumor samples of melanoma patients treated with checkpoint inhibitors. Two distinct states of CD8(+) T cells were defined by clustering and associated with patient tumor regression or progression. A single transcription factor, TCF7, was visualized within CD8(+) T cells in fixed tumor samples and predicted positive clinical outcome in an independent cohort of checkpoint-treated patients. We delineated the epigenetic landscape and clonality of these T cell states and demonstrated enhanced antitumor immunity by targeting novel combinations of factors in exhausted cells. Our study of immune cell transcriptomes from tumors demonstrates a strategy for identifying predictors, mechanisms, and targets for enhancing checkpoint immunotherapy.
-
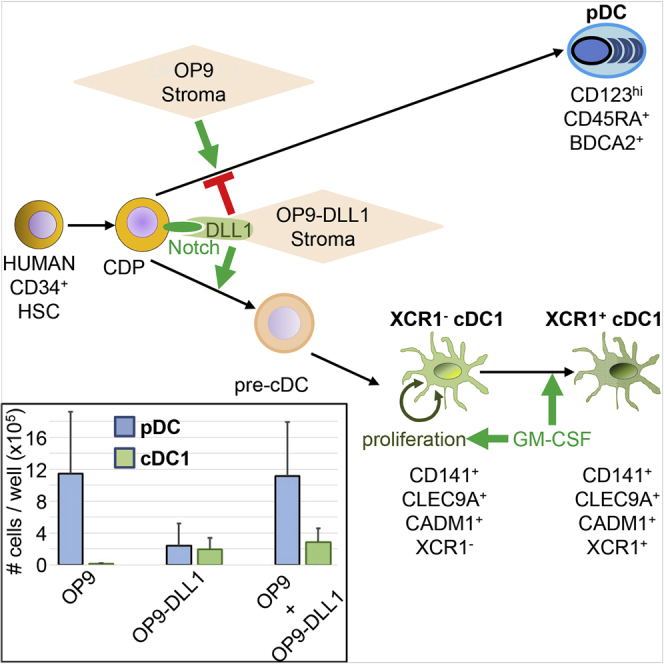
Large-Scale Human Dendritic Cell Differentiation Revealing Notch-Dependent Lineage Bifurcation and Heterogeneity
Cell Rep, 24(7):1902-1915.e6. 2018 Aug 14.
Abstract
The ability to generate large numbers of distinct types of human dendritic cells (DCs) in vitro is critical for accelerating our understanding of DC biology and harnessing them clinically. We developed a DC differentiation method from human CD34(+) precursors leading to high yields of plasmacytoid DCs (pDCs) and both types of conventional DCs (cDC1s and cDC2s). The identity of the cells generated in vitro and their strong homology to their blood counterparts were demonstrated by phenotypic, functional, and single-cell RNA-sequencing analyses. This culture system revealed a critical role of Notch signaling and GM-CSF for promoting cDC1 generation. Moreover, we discovered a pre-terminal differentiation state for each DC type, characterized by high expression of cell-cycle genes and lack of XCR1 in the case of cDC1. Our culture system will greatly facilitate the simultaneous and comprehensive study of primary, otherwise rare human DC types, including their mutual interactions.
-
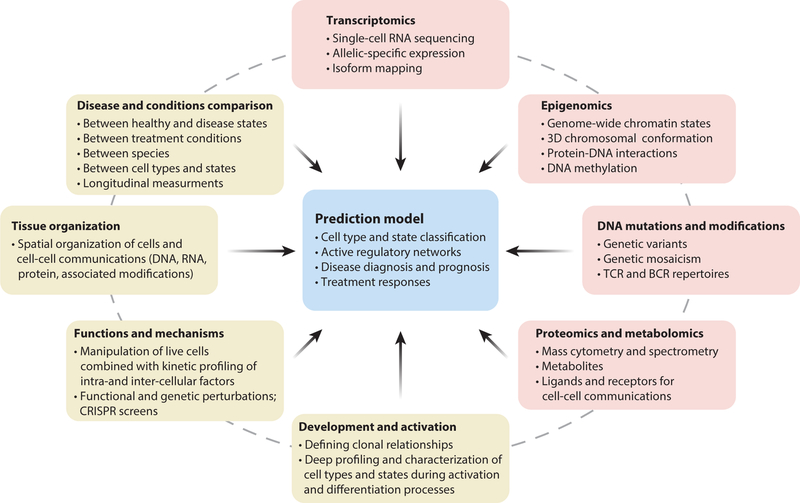
Systems Immunology: Learning the Rules of the Immune System
Annu Rev Immunol, 36:813-842. 2018 Apr 26.
Abstract
Given the many cell types and molecular components of the human immune system, along with vast variations across individuals, how should we go about developing causal and predictive explanations of immunity? A central strategy in human studies is to leverage natural variation to find relationships among variables, including DNA variants, epigenetic states, immune phenotypes, clinical descriptors, and others. Here, we focus on how natural variation is used to find patterns, infer principles, and develop predictive models for two areas: (a) immune cell activation-how single-cell profiling boosts our ability to discover immune cell types and states-and (b) antigen presentation and recognition-how models can be generated to predict presentation of antigens on MHC molecules and their detection by T cell receptors. These are two examples of a shift in how we find the drivers and targets of immunity, especially in the human system in the context of health and disease.
-
-
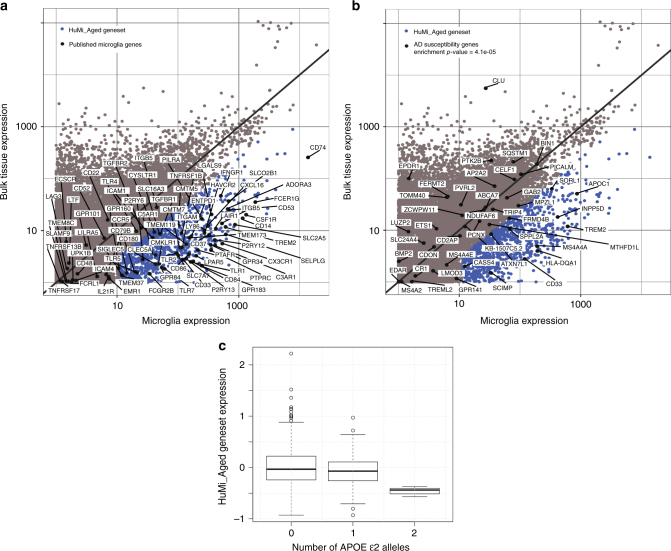
A transcriptomic atlas of aged human microglia
Nat Commun, 9(1):539. 2018 Feb 7.
Abstract
With a rapidly aging global human population, finding a cure for late onset neurodegenerative diseases has become an urgent enterprise. However, these efforts are hindered by the lack of understanding of what constitutes the phenotype of aged human microglia-the cell type that has been strongly implicated by genetic studies in the pathogenesis of age-related neurodegenerative disease. Here, we establish the set of genes that is preferentially expressed by microglia in the aged human brain. This HuMi_Aged gene set captures a unique phenotype, which we confirm at the protein level. Furthermore, we find this gene set to be enriched in susceptibility genes for Alzheimer's disease and multiple sclerosis, to be increased with advancing age, and to be reduced by the protective APOEε2 haplotype. APOEε4 has no effect. These findings confirm the existence of an aging-related microglial phenotype in the aged human brain and its involvement in the pathological processes associated with brain aging.
2017 (4)
-
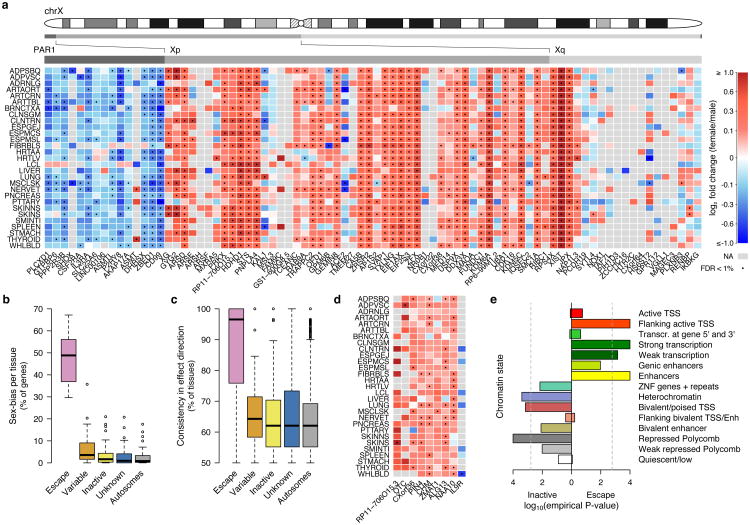
Landscape of X chromosome inactivation across human tissues
Nature, 550(7675):244-248. 2017 Oct 11.
Abstract
X chromosome inactivation (XCI) silences transcription from one of the two X chromosomes in female mammalian cells to balance expression dosage between XX females and XY males. XCI is, however, incomplete in humans: up to one-third of X-chromosomal genes are expressed from both the active and inactive X chromosomes (Xa and Xi, respectively) in female cells, with the degree of 'escape' from inactivation varying between genes and individuals. The extent to which XCI is shared between cells and tissues remains poorly characterized, as does the degree to which incomplete XCI manifests as detectable sex differences in gene expression and phenotypic traits. Here we describe a systematic survey of XCI, integrating over 5,500 transcriptomes from 449 individuals spanning 29 tissues from GTEx (v6p release) and 940 single-cell transcriptomes, combined with genomic sequence data. We show that XCI at 683 X-chromosomal genes is generally uniform across human tissues, but identify examples of heterogeneity between tissues, individuals and cells. We show that incomplete XCI affects at least 23% of X-chromosomal genes, identify seven genes that escape XCI with support from multiple lines of evidence and demonstrate that escape from XCI results in sex biases in gene expression, establishing incomplete XCI as a mechanism that is likely to introduce phenotypic diversity. Overall, this updated catalogue of XCI across human tissues helps to increase our understanding of the extent and impact of the incompleteness in the maintenance of XCI.
-
Aryl Hydrocarbon Receptor Controls Monocyte Differentiation into Dendritic Cells versus Macrophages
Immunity, 47(3):582-596.e6. 2017 Sep 19.
Abstract
After entering tissues, monocytes differentiate into cells that share functional features with either macrophages or dendritic cells (DCs). How monocyte fate is directed toward monocyte-derived macrophages (mo-Macs) or monocyte-derived DCs (mo-DCs) and which transcription factors control these differentiation pathways remains unknown. Using an in vitro culture model yielding human mo-DCs and mo-Macs closely resembling those found in vivo in ascites, we show that IRF4 and MAFB were critical regulators of monocyte differentiation into mo-DCs and mo-Macs, respectively. Activation of the aryl hydrocarbon receptor (AHR) promoted mo-DC differentiation through the induction of BLIMP-1, while impairing differentiation into mo-Macs. AhR deficiency also impaired the in vivo differentiation of mouse mo-DCs. Finally, AHR activation correlated with mo-DC infiltration in leprosy lesions. These results establish that mo-DCs and mo-Macs are controlled by distinct transcription factors and show that AHR acts as a molecular switch for monocyte fate specification in response to micro-environmental factors.
-
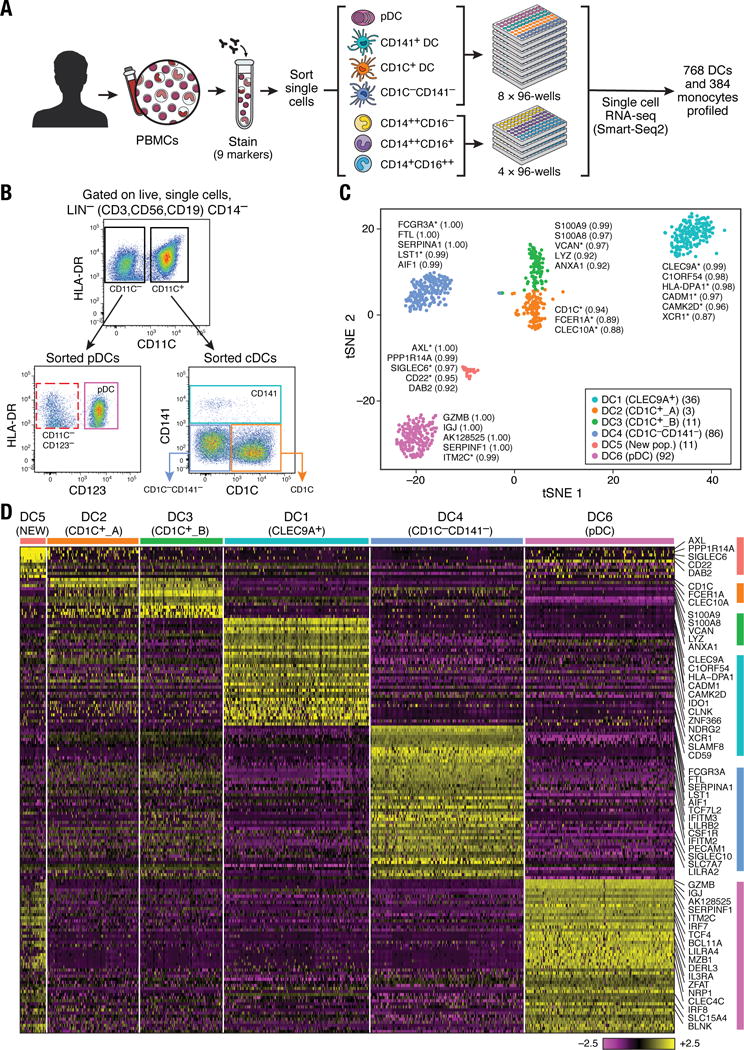
Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors
Science, 356(6335). 2017 Apr 21.
Abstract
Dendritic cells (DCs) and monocytes play a central role in pathogen sensing, phagocytosis, and antigen presentation and consist of multiple specialized subtypes. However, their identities and interrelationships are not fully understood. Using unbiased single-cell RNA sequencing (RNA-seq) of ~2400 cells, we identified six human DCs and four monocyte subtypes in human blood. Our study reveals a new DC subset that shares properties with plasmacytoid DCs (pDCs) but potently activates T cells, thus redefining pDCs; a new subdivision within the CD1C(+) subset of DCs; the relationship between blastic plasmacytoid DC neoplasia cells and healthy DCs; and circulating progenitor of conventional DCs (cDCs). Our revised taxonomy will enable more accurate functional and developmental analyses as well as immune monitoring in health and disease.
-
Single-Cell RNA Sequencing of Human T Cells
Methods Mol Biol, 1514:203-239. 2017.
Abstract
Understanding how populations of human T cells leverage cellular heterogeneity, plasticity, and diversity to achieve a wide range of functional flexibility, particularly during dynamic processes such as development, differentiation, and antigenic response, is a core challenge that is well suited for single-cell analysis. Hypothesis-free evaluation of cellular states and subpopulations by transcriptional profiling of single T cells can identify relationships that may be obscured by targeted approaches such as FACS sorting on cell-surface antigens, or bulk expression analysis. While this approach is relevant to all cell types, it is of particular interest in the study of T cells for which classical phenotypic criteria are now viewed as insufficient for distinguishing different T cell subtypes and transitional states, and defining the changes associated with dysfunctional T cell states in autoimmunity and tumor-related exhaustion. This unit describes a protocol to generate single-cell transcriptomic libraries of human blood CD4(+) and CD8(+) T cells, and also introduces the basic bioinformatic steps to process the resulting sequence data for further computational analysis. We show how cellular subpopulations can be identified from transcriptional data, and derive characteristic gene expression signatures that distinguish these states. We believe single-cell RNA-seq is a powerful technique to study the cellular heterogeneity in complex tissues, a paradigm that will be of great value for the immune system.
2016 (1)
-
Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq
Science, 352(6282):189-96. 2016 Apr 8.
Abstract
To explore the distinct genotypic and phenotypic states of melanoma tumors, we applied single-cell RNA sequencing (RNA-seq) to 4645 single cells isolated from 19 patients, profiling malignant, immune, stromal, and endothelial cells. Malignant cells within the same tumor displayed transcriptional heterogeneity associated with the cell cycle, spatial context, and a drug-resistance program. In particular, all tumors harbored malignant cells from two distinct transcriptional cell states, such that tumors characterized by high levels of the MITF transcription factor also contained cells with low MITF and elevated levels of the AXL kinase. Single-cell analyses suggested distinct tumor microenvironmental patterns, including cell-to-cell interactions. Analysis of tumor-infiltrating T cells revealed exhaustion programs, their connection to T cell activation and clonal expansion, and their variability across patients. Overall, we begin to unravel the cellular ecosystem of tumors and how single-cell genomics offers insights with implications for both targeted and immune therapies.
2015 (1)
-
Sex Differences in Plasmacytoid Dendritic Cell Levels of IRF5 Drive Higher IFN-α Production in Women
J Immunol, 195(11):5327-36. 2015 Dec 1.
Abstract
Increased IFN-α production contributes to the pathogenesis of infectious and autoimmune diseases. Plasmacytoid dendritic cells (pDCs) from females produce more IFN-α upon TLR7 stimulation than pDCs from males, yet the mechanisms underlying this difference remain unclear. In this article, we show that basal levels of IFN regulatory factor (IRF) 5 in pDCs were significantly higher in females compared with males and positively correlated with the percentage of IFN-α-secreting pDCs. Delivery of recombinant IRF5 protein into human primary pDCs increased TLR7-mediated IFN-α secretion. In mice, genetic ablation of the estrogen receptor 1 (Esr1) gene in the hematopoietic compartment or DC lineage reduced Irf5 mRNA expression in pDCs and IFN-α production. IRF5 mRNA levels furthermore correlated with ESR1 mRNA levels in human pDCs, consistent with IRF5 regulation at the transcriptional level by ESR1. Taken together, these data demonstrate a critical mechanism by which sex differences in basal pDC IRF5 expression lead to higher IFN-α production upon TLR7 stimulation in females and provide novel targets for the modulation of immune responses and inflammation.
2014 (1)
-
Common genetic variants modulate pathogen-sensing responses in human dendritic cells
Science, 343(6175):1246980. 2014 Mar 7.
Abstract
Little is known about how human genetic variation affects the responses to environmental stimuli in the context of complex diseases. Experimental and computational approaches were applied to determine the effects of genetic variation on the induction of pathogen-responsive genes in human dendritic cells. We identified 121 common genetic variants associated in cis with variation in expression responses to Escherichia coli lipopolysaccharide, influenza, or interferon-β (IFN-β). We localized and validated causal variants to binding sites of pathogen-activated STAT (signal transducer and activator of transcription) and IRF (IFN-regulatory factor) transcription factors. We also identified a common variant in IRF7 that is associated in trans with type I IFN induction in response to influenza infection. Our results reveal common alleles that explain interindividual variation in pathogen sensing and provide functional annotation for genetic variants that alter susceptibility to inflammatory diseases.
2013 (1)
-
Identification of regulators of the innate immune response to cytosolic DNA and retroviral infection by an integrative approach
Nat Immunol, 14(2):179-85. 2013 Feb.
Abstract
The innate immune system senses viral DNA that enters mammalian cells, or in aberrant situations self-DNA, and triggers type I interferon production. Here we present an integrative approach that combines quantitative proteomics, genomics and small molecule perturbations to identify genes involved in this pathway. We silenced 809 candidate genes, measured the response to dsDNA and connected resulting hits with the known signaling network. We identified ABCF1 as a critical protein that associates with dsDNA and the DNA-sensing components HMGB2 and IFI204. We also found that CDC37 regulates the stability of the signaling molecule TBK1 and that chemical inhibition of the CDC37-HSP90 interaction and several other pathway regulators potently modulates the innate immune response to DNA and retroviral infection.
2010 (2)
-
Genetic risk factors for post-infectious irritable bowel syndrome following a waterborne outbreak of gastroenteritis
Gastroenterology, 138(4):1502-13. 2010 Apr.
Abstract
BACKGROUND & AIMS: Acute gastroenteritis is the strongest risk factor for irritable bowel syndrome (IBS). In May 2000, >2300 residents of Walkerton, Ontario, developed gastroenteritis from microbial contamination of the municipal water supply; a longitudinal study found that >36.2% of these developed IBS. We used this cohort to study genetic susceptibility to post-infectious (PI)-IBS. METHODS: We screened 79 functional variants of genes with products involved in serotoninergic pathways, intestinal epithelial barrier function, and innate immunity and performed fine mapping in regions of interest. We compared data from Walkerton residents who developed gastroenteritis and reported PI-IBS 2 to 3 years after the outbreak (n = 228, cases) with data from residents who developed gastroenteritis but did not develop PI-IBS (n = 581, controls). RESULTS: Four variants were associated with PI-IBS, although the association was not significant after correction for the total number of single nucleotide polymorphisms. Two were located in TLR9, which encodes a pattern recognition receptor (rs352139, P545P; P = .0059 and rs5743836, -T1237C; P = .0250; r(2) < 0.14); 1 was in CDH1, which encodes a tight junction protein (rs16260, -C160A; P = .0352); and 1 was in IL6, which encodes a cytokine (rs1800795, -G174C; P = .0420). Denser mapping of these 3 regions revealed 1 novel association in IL6 (rs2069861; P = .0069) and 14 associations that could be accounted for by linkage disequilibrium with the 4 original variants. The TLR9, IL6, and CDH1 variants all persisted as independent risk factors for PI-IBS when controlling for previously identified clinical risk factors. CONCLUSION: This is the first descriptive study to assess potential genetic determinants of PI-IBS. Genes that encode proteins involved in epithelial cell barrier function and the innate immune response to enteric bacteria are associated with development of IBS following acute gastroenteritis.
-
[Bacterial recognition of the intestinal microbial flora: an important risk factor for Crohn's disease]
Med Sci (Paris), 26(1):36-7. 2010 Jan.
2009 (3)
-
Genetic variation in the familial Mediterranean fever gene (MEFV) and risk for Crohn's disease and ulcerative colitis
PLoS One, 4(9):e7154. 2009 Sep 28.
Abstract
BACKGROUND AND AIMS: The familial Mediterranean fever (FMF) gene (MEFV) encodes pyrin, a major regulator of the inflammasome platform controlling caspase-1 activation and IL-1beta processing. Pyrin has been shown to interact with the gene product of NLRP3, NALP3/cryopyrin, also an important active member of the inflammasome. The NLRP3 region was recently reported to be associated with Crohn's disease (CD) susceptibility. We therefore sought to evaluate MEFV as an inflammatory bowel disease (IBD) susceptibility gene. METHODOLOGY AND RESULTS: MEFV colonic mucosal gene expression was significantly increased in experimental colitis mice models (TNBS p<0.0003; DSS p<0.006), in biopsies from CD (p<0.02) and severe ulcerative colitis (UC) patients (p<0.008). Comprehensive genetic screening of the MEFV region in the Belgian exploratory sample set (440 CD trios, 137 UC trios, 239 CD cases, 96 UC cases, and 107 healthy controls) identified SNPs located in the MEFV 5' haplotype block that were significantly associated with UC (rs224217; p = 0.003; A allele frequency: 56% cases, 45% controls), while no CD associations were observed. Sequencing and subsequent genotyping of variants located in this associated haplotype block identified three synonymous variants (D102D/rs224225, G138G/rs224224, A165A/rs224223) and one non-synonymous variant (R202Q/rs224222) located in MEFV exon 2 that were significantly associated with UC (rs224222: p = 0.0005; A allele frequency: 32% in cases, 23% in controls). No consistent associations were observed in additional Canadian (256 CD trios, 91 UC trios) and Scottish (495 UC, 370 controls) sample sets. We note that rs224222 showed marginal association (p = 0.012; G allele frequency: 82% in cases, 70% in controls) in the Canadian sample, but with a different risk allele. None of the NLRP3 common variants were associated with UC in the Belgian-Canadian UC samples and no significant interactions were observed between NLRP3 and MEFV that could explain the observed flip-flop of the rs224222 risk allele. CONCLUSION: The differences in association levels observed between the sample sets may be a consequence of distinct founder effects or of the relative small sample size of the cohorts evaluated in this study. However, the results suggest that common variants in the MEFV region do not contribute to CD and UC susceptibility.
-
The contribution of genetic studies in shifting the immunopathogenesis paradigm of Crohn's disease
Expert Rev Clin Immunol, 5(4):361-4. 2009 Jul.
-
Common variants in the NLRP3 region contribute to Crohn's disease susceptibility
Nat Genet, 41(1):71-6. 2009 Jan.
Abstract
We used a candidate gene approach to identify a set of SNPs, located in a predicted regulatory region on chromosome 1q44 downstream of NLRP3 (previously known as CIAS1 and NALP3) that are associated with Crohn's disease. The associations were consistently replicated in four sample sets from individuals of European descent. In the combined analysis of all samples (710 father-mother-child trios, 239 cases and 107 controls), these SNPs were strongly associated with risk of Crohn's disease (P(combined) = 3.49 x 10(-9), odds ratio = 1.78, confidence interval = 1.47-2.16 for rs10733113), reaching a level consistent with the stringent significance thresholds imposed by whole-genome association studies. In addition, we observed significant associations between SNPs in the associated regions and NLRP3 expression and IL-1beta production. Mutations in NLRP3 are known to be responsible for three rare autoinflammatory disorders. These results suggest that the NLRP3 region is also implicated in the susceptibility of more common inflammatory diseases such as Crohn's disease.
2005 (1)
-
Tumor necrosis factor receptor gene polymorphisms in Crohn's disease: association with clinical phenotypes
Am J Gastroenterol, 100(5):1126-33. 2005 May.
Abstract
OBJECTIVES: Crohn's disease (CD) is a chronic multifactorial disorder with diverse clinical features that are influenced by a heterogeneous set of genetic factors. TNF-alpha/TNF receptor interactions play a pivotal role in the pathogenesis of the inflammatory response. Our purpose was to determine whether single nucleotide polymorphisms (SNPs) in the TNF receptors confer susceptibility to Crohn's disease and whether they are associated with clinical phenotype. METHODS: A cohort of 205 consecutively identified and unrelated patients with CD and 106 controls were recruited. Subjects were genotyped for polymorphisms in TNFRSF1A (position +36, -609), TNFRSF1B (+196, +1466), along with the three common CARD15 variants and phenotyped for disease behavior. Genotypic and allelic frequencies were compared between CD and controls and a logistic regression model was constructed to determine independent associations with specific clinical phenotypes. RESULTS: Only the TNFRSF1A +36 and TNFRSF1B +196 SNPs were associated with CD (p= 0.0019 and 0.034, respectively). The TNFRSF1A +36 mutation was negatively associated with stricturing disease phenotype (OR = 0.384; CI = 0.166-0.887). In contrast, the TNFRSF1B +196 was negatively associated with colitis (OR = 0.410; CI = 0.191-0.880). These associations were independent of CARD15 mutation status. Finally, TNFRSF1B +196 was negatively associated with surgery in CARD15 negative patients. CONCLUSIONS: These data constitute the first report of an association of TNFRSF1A and TNFRSF1B polymorphisms with CD in a Caucasian population and address the role of TNFR mutations in determining clinical heterogeneity in CD.